Citation:
Si ZHOU, Yan-Yan ZHAO, Ji-Jun ZHAO. Cage Clusters: from Structure Prediction to Rational Design of Functional Nanomaterials[J]. Chinese Journal of Structural Chemistry,
2020, 39(7): 1185-1193.
doi:
10.14102/j.cnki.0254–5861.2011–2922
Cage Clusters: from Structure Prediction to Rational Design of Functional Nanomaterials
Received Date:
28 June 2020 Accepted Date:
05 July 2020 Available Online:
01 July 2020
Fund Project:
Abstract:
Atomic clusters of subnanometer scale and variable chemical composition offer great opportunities for rational design of functional nanomaterials. Among them, cage clusters doped with endohedral atom are particularly interesting owing to their enhanced stability and highly tunable physical and chemical properties. In this perspective, first we give a brief overview of the history of doped cage clusters and introduce the home-developed comprehensive genetic algorithm (CGA) for structure prediction of clusters. Then, we show a few examples of magnetic clusters and subnanometer catalysts based on doped cage clusters, which are computationally revealed or designed by the CGA code. Finally, we give an outlook for some future directions of cluster science.
As aggregates of a few to hundreds of atoms, clusters are considered as an intermediate stage between the microscopic atoms/molecules and the macroscopic condensed matters. The advantage of utilizing clusters as building blocks of functional nanomaterials lies in the following two aspects: (ⅰ) The composition and size of an atomic cluster are continuously tunable, enabling the precise modulation of electronic structure, (ⅱ) Most atoms in the cluster are on the surface, thereby leading to superior chemical activity. To serve as a building block, a cluster should have desirable physical and chemical properties, as well as sufficiently high stability for retaining its identity in the cluster-assembled materials and devices. Among atomic clusters with various morphologies, cage clusters usually doped with one or several heteroatoms offer great possibilities to meet the above criteria.
The original idea of doped cage clusters stemmed from a pioneer study by Gong and Kumar in 1993[1]. By doping a group 14 atom X (X = C, Si, and Ge) into the cavity of Al12 icosahedron, the resulting X@Al12 clusters attach 40 valence electrons to reach the closed electronic shell within a spherical jellium picture[2] and are thus remarkably stable. Following the famous "18-electron principle"[3], in 2002 Pyykkö and Rune- berg theoretically predicted a similar icosahedral W@Au12 cluster with high stability and large HOMO- LUMO gap[4], which was experimentally confirmed by the photoelectron spectrum from Wang's group[5].
Analogous to endohedral carbon fullerenes[6], Kumar and Kawazoe proposed to dope a group 4 transition metal atom (Ti, Zr, Hf) into a Si16 cage[7] or Ge16 cage[8], which not only stabilizes the fullerene- like or Frank-Kasper (FK) type structure, but also leads to closed electronic shell with totally 68 valence electrons. Later, the cage geometry, high stability and large HOMO-LUMO gap of these M@Si16 (or M@Ge16) clusters and their isoelectronic counterparts have been verified using experimental mass spectrometry, photoelectron spectroscopy, and X-ray spectroscopy[9-11]. Recently, Ta@Si16 and Ti@Si16 clusters up to 100 mg in amount have been produced by a dry-wet combination synthesis method, paving an avenue towards cluster assemblies built from these highly stable cage clusters[12].
During the past two decades, there have been enormous theoretical and experimental studies on the endohedrally doped cage clusters[13]; however, most of them have been devoted to the atomic structures and fundamental properties, and there is a lack of rational design of functional materials based on these clusters. In this perspective, we highlight a few examples of doped cage clusters from our recent studies[14-22]. Cage structures of transition metal doped Si, Ge, and Au clusters have been determined from global search using our comprehensive genetic algorithm (CGA) code and ab initio calculations[23], possibly with the aid of experimental photoelectron spectroscopic data. These cage clusters were found to possess intriguing magnetic behavior, from ferromagnetic to antiferromagnetic and ferrimagnetic. Starting from these cage clusters, subnanometer catalysts have been further designed for CO oxidation and CO2 reduction. All these results demonstrate that doped cage clusters are promising candidates to build functional nanomaterials, and the state-of-the-arts structure prediction methods provide powerful tools for discovering novel clusters with desirable physical and chemical properties.
2.
COMPREHENSIVE GENETIC ALGORITHM FOR STRUCTURE PREDICTION OF CLUSTERS
In cluster science, theoretical study of a cluster starts from determination of its ground state configuration. To this end, one has to conduct an extensive search on the potential energy surface (PES) to locate the global minimum, which is challenging due to the extremely complicated nature of the PES for a cluster (especially those with more than 20 atoms). Evidently, some efficient and robust algorithms for global optimization are needed. Inspired by the Darwin's theory of evolution that only the fittest individual can survive, Holland introduced genetic algorithm (GA) into computer science in 1960s and his student Goldberg further extended GA in 1989[24]. Briefly speaking, genetic algorithm is an evolutional process of natural selection of a population using some biologically inspired operators such as mutation, crossover and selection. In the mid-1990s, GA was extended to cluster science, as demonstrated by the success in obtaining the C60 buckyball structure from scratch[25]. Since our first paper about (C60)n molecular clusters with n = 2~25 in 1999[26], we have been developing GA for structure prediction and applied it to a large variety of clusters[27] as well as selected atomic wires[28]. In 2016, we put forward an upgrade version of our GA code and named it as comprehensive genetic algorithm (CGA)[23].
In practice, CGA can be combined with either density functional theory (DFT) codes (like DMol3 and VASP) or empirical molecular dynamics codes (like LAMMPS) to sample the PES[29-31]. In the CGA search, a number of random configurations were generated as the initial population. At each step of CGA iteration, any two individuals in the population were selected as parents to produce a child cluster via a "cut and splice" mating operation[25], followed by an optional mutation operation and a local geometry optimization on the child cluster. Special operations on the cluster geometries have been developed, such as principle direction mating, radial direction mating, and operation within symmetry constraint. Typical mutation operations include: (i) applying a small random displacement on each atom, and (ii) exchanging the elements of two atoms. In order to ensure the diversity of the populations and to avoid identical geometries in the population, we used the mass moment of inertia to distinguish different isomers. The number of CGA iterations and size of population generally increase with cluster size, and rely on the specific chemical composition of the cluster. The high efficiency of CGA-DFT scheme has been well demonstrated by its applications to medium-sized clusters (c.a. 50 atoms), such as B48[32], Na59+[33], and (WO3)12[34]. More details about CGA can be found in our earlier review article[23]. Using CGA-DFT global search, we have unveiled a series of doped cage clusters, and some of them will be discussed in the following contents.
3.
MAGNETIC CLUSTERS OF DOPED SILICON AND GERMANIUM CAGES
The magnetic properties of clusters and the regulation for tailoring the magnetism of cluster assemblies are scientifi-cally interesting and significant for technological applications[35]. Kumar and Kawazoe[36] proposed a generic way to find stable magnetic cluster by doping a transition metal atom (e.g., Mn) into a cluster of group 14 element (Si, Ge, Sn). According to Hund's rule, many transition metal atoms with half-filled d shell (especially the 3d series) possess a magnetic moment. It is thus interesting to explore whether such magnetic moment can be retained when the transition metal atom is doped into a Si or Ge host cluster. Using DFT calculations, our group investigated the evolution behavior of magnetic properties of Fe-doped Sin clusters with n = 2~14[37]. For smaller sizes (n = 2~8), the total magnetic moment of FeSin cluster is 2 µB and mainly located on the Fe site. However, the magnetic moment of Fe atom in larger FeSin clusters with n = 9 and 11~14 is completely quenched except FeSi10 having a magnetic moment of about 1 µB. The quenching of the magnetic moment of Fe atom was attributed to the significant charge transfer and strong hybridization between Fe 4s, 3d and Si 3s, 3p states. Similar size-dependent quenching of magnetic moment was also observed in doped CrSin−[38] and MnGen[39] clusters.
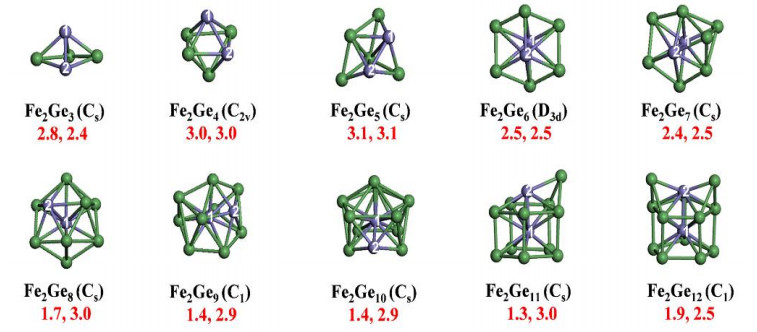
To avoid magnetic quenching that typically occurs in single transition metal atom doped Si (Ge) clusters, we explored the magnetic properties of dual Fe atoms doped Gen−/0 (n = 3~12) clusters using CGA-DFT search[18]. First of all, the ground state structures and associated electronic states of anionic Fe2Gen− clusters were confirmed by experimental photoelectron spectra of our own. The lowest-energy structures and on-site magnetic moments of neutral Fe2Gen clusters are shown in Fig. 1. Starting from Fe2Ge9, endohedral cage structure emerges with one Fe atom staying in the cage center and the other locating on the cage surface. Further adding Ge atoms leads to capped endohedral cage configurations for Fe2Ge11 and Fe2Ge12. Interestingly, the on-site magnetic moments of the two Fe atoms exhibit ferromagnetic coupling with total magnetic moment of 4 μB for most clusters and 6 μB for Fe2Ge4 and Fe2Ge5, largely preserved as compared to a free Fe2 dimer (6 μB). The variation of spin moments of Fe2Gen mainly stems from the charge transfer between the Fe atoms and host Gen cluster, i.e., the fewer electrons Fe atoms gain from Gen cage, the larger spin moment they carry on.
Figure 1
Figure 1.
The lowest-energy structures of Fe2Gen (n = 3~12) clusters. The symmetries are given in parentheses. The red numbers (left and right) below each structure show magnetic moments (in unit of µB) for Fe1 and Fe2 atoms, respectively. Green and purple balls represent Ge and Fe atoms, respectively[18]
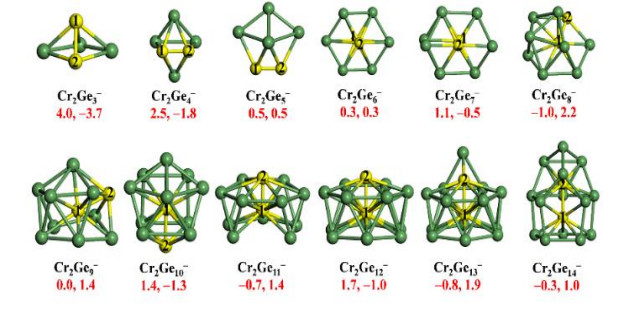
It is known that bulk Cr metal is antiferromagnetic, and hence it is an intriguing question if the antiferromagnetism can be retained for a Cr dimer doped in the germanium clusters. In a subsequent study, we explored the structures and magnetic properties of anionic Cr2Gen− (n = 3~14) clusters[19]. The lowest-energy structures and on-site magnetic moments of Cr2Gen− are shown in Fig. 2. The agreement between theoretical and experimental photoelectron spectra supports the assignment of ground state structures for Cr2Gen− by the CGA search. Again, Cr-centered cage structure starts to form at Cr2Ge9−. A noticeable cage cluster with C2v symmetry was obtained for Cr2Ge12−, which can be viewed as a capped hexagonal antiprism with an endohedral Cr atom. Unlike dual Fe atoms doped Ge clusters, most Cr2Gen− (n = 3~14) clusters exhibit antiferromagnetic Cr–Cr coupling, except for Cr2Ge5− and Cr2Ge6− with ferromagnetic coupling. All these clusters have a total magnetic moment of 1 μB, which is contributed not only by the Cr dopants, but also by the host Gen cage. The antiferromagnetism was attributed to the different coordination environment of the two Cr atoms in the cage, which results in distinct occupancies of their d orbitals and p-d coupling strengths between Cr and Ge atoms for the interior and exterior Cr dopants.
Figure 2
Figure 2.
The lowest-energy structures of Cr2Gen− (n = 3~14) clusters. The red numbers (left and right) below each structure show magnetic moments (in unit of µB) for Cr1 and Cr2 atoms, respectively. Green and yellow balls represent Ge and Cr atoms, respectively[19]
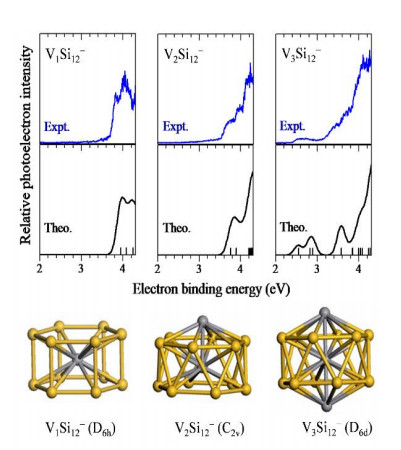
Would doping of more metal atoms alter the magnetic ordering of cage clusters? Fig. 3 presents the photoelectron spectra and ground state structures of VxSi12− (x = 1, 2, 3) clusters from our combined CGA-DFT and photoelectron spectroscopic study[14]. Endohedral cages structures were found for all the three doped clusters, that is, V@Si12− is a Vcentered hexagonal prism, V2Si12− is a capped hexagonal antiprism similar to Cr2Ge12−, and V3Si12− is a bicapped hexagonal antiprism. The total magnetic moments are 0, 1 and 4 μB for V@Si12−, V2Si12− and V3Si12−, respectively. Among them, a particularly interesting species is the wheel-like V3Si12−, in which the on-site magnetic moments on V atoms are ferrimagnetic with +2.4 μB on each of the surface V atoms and −0.6 μB on the interior V atom. Similar to the case of Cr2@Gen−, the variation of chemical environment (coordination numbers and V–Si bond length) of three V atoms induces distinct p-d hybridization between the V and Si atoms, and thus endows different local magnetic moments on the V atoms. It is also noteworthy that V3Si12− is the only magnetic species among all the V3Sin− clusters within the size range of n = 3~14[15].
Figure 3
Figure 3.
Photoelectron spectra (upper) from experiment and theoretical calculation, and ground state structures (lower) of VxSi12− (x = 1, 2, 3) clusters[14]
These series of studies show that doping single or multiple transition metal atoms in Si (Ge) clusters can not only prevent the magnetic quenching effect, but also induce different magnetic coupling between the metal dopants, including ferromagnetic, antiferromagnetic and ferrimagnetic. The highly tunable magnetic behavior of these metal doped silicon and germanium clusters make them promising building block for nanoscale spintronics and high-density magnetic storage.
4.
SUBNANOMETER CATALYSTS OF DOPED SILICON, GERMANIUM, AND GOLD CAGES
The electronic structures and surface properties of endohedral cage clusters can be effectively tailored by both cluster size and dopant atom, rendering them a family of promising catalysts for energy and environmental applications. Recently, we have made some theoretical efforts in exploring the activity of endohedrally doped clusters for various chemical reactions.
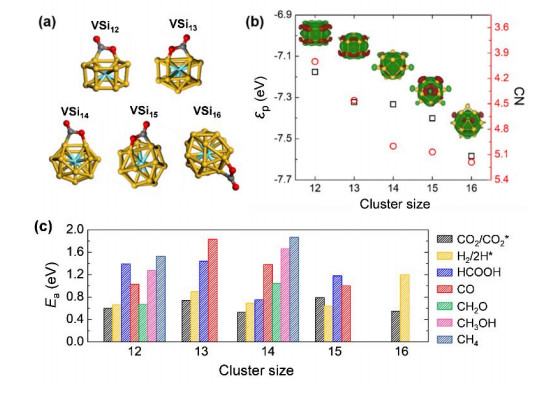
A previous study using infrared multiple photon dissociation spectroscopy showed that V-doped Sin clusters adopt endohedral cages in size range of n = 12~16[40], while our combined CGA-DFT and photoelectron spectroscopic study also supported the cage structure for V@Si12[14]. Then, these V@Sin cage clusters were proposed for photocatalysis of CO2 hydrogenation based on our DFT calculations[17]. As shown in Fig. 4, we demonstrated that the vanadium-doped silicon cages carry unsaturated states and can activate CO2 molecules. Hydrogenation of CO2 on V@Sin leads to the formation of CO, formic acid, formaldehyde, methanol, and methane with barriers down to 0.67~1.53 eV, and the product selectivity is uniquely determined by the cluster size. The suitable HOMO-LUMO gap and high visible light absorption of the V@Sin clusters can further drive the catalysis. Moreover, the CO2 adsorption strength of these silicon cages is correlated to the p orbital center of Si atoms, which is in turn mediated by the charge transfer between V and Si atoms. Such electronic structure-activity relationship conforms to a recently established "p band theory" for non-metal catalysts[41-43], which provides the guidelines for catalysts design using those earth abundant p-block elements.
Figure 4
Figure 4.
(a) Structures of V@Sin (n = 12~16) clusters chemisorbed with a CO2 molecule, with C, O, Si, and V atoms represented in gray, red, yellow, and cyan colors, respectively. (b) The p orbital center (εp, black symbols) and coordination number (CN, red symbols) of Si atoms as a function of the size of V@Sin clusters. The insets display the differential charge density distributions between the V atom and Si cage. The electron accumulation and depletion regions are represented by red and green colors, respectively, using an isosurface value of 2 × 10−3 e/Å3. (c) Kinetic barrier (Ea) for chemisorption of CO2 and dissociation of the H2 molecule, and the barrier of the rate-limiting step for CO2 hydrogenation to various products on the V@Sin clusters[17]
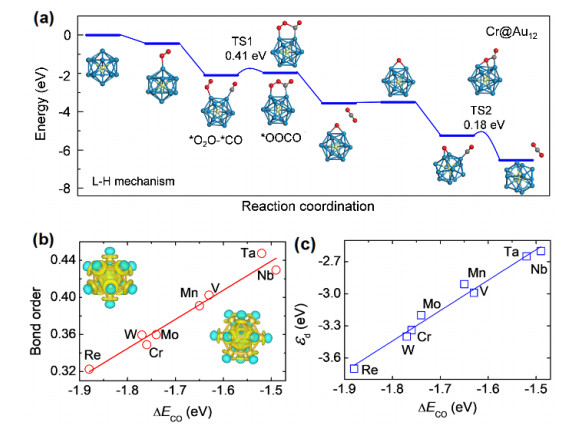
The effect of endohedral doping on the activity of metal clusters has also been investigated by our group. Recently, we have performed systematical CGA-DFT search on the PES of M@Au12 (M = 3d~5d elements) clusters[21]. Among them, the ground state structures of Mo@Au12 or W@Au12 clusters is the perfect icosahedron cage, the M@Au12 (M = V, Nb, Ta, Tc, and Re) clusters adopt the distorted icosahedron cages, and the lowest-energy structures of M@Au12 (M = Sc, Ti, Cr, Mn, Fe, Co, Ru, Rh, and Ir) are the perfect or distorted magnetic cuboctahedron cages. Furthermore, we considered various transition metal (M) doped Au12 cage clusters for CO oxidation with M = V, Cr, Mn, Nb, Mo, Ta, W, and Re[22], and the essential results are summarized in Fig. 5. Our DFT calculations revealed prominent charge densities on the cage surface, which are related to the electronic states near HOMO and responsible for the activity. The adsorption strength of CO, O2 and intermediate species on the M@Au12 clusters is highly sensitive to the doping element, i.e. it follows linear correlation to the M–Au bond order and d orbital level of M@Au12. The highest activity was found for Cr@Au12 and Mn@Au12 that provide suitable binding strength and have reaction barriers of only about 0.41 eV for CO oxidation under the Langumir-Hinshelwood mecha- nism.
Figure 5
Figure 5.
(a) Reaction pathways of CO oxidation on the Cr@Au12 cluster under the L-H mechanism. The insets show the structures of the corresponding reaction intermediates and transition states (TS). The numbers indicate the kinetic barriers. The C, O, Cr and Au atoms are shown in grey, red, green and blue colors, respectively. (b) M−Au bond order and (c) d orbital center (εd) as a function of CO adsorption energy for various M@Au12 clusters. The insets in (b) show the differential charge density distributions of Cr@Au12 (Ih) and Ta@Au12 (Oh). The yellow and cyan colors represent the electron accumulation and depletation regions with isosurface value of 0.015 e/Å3, respectively[22]
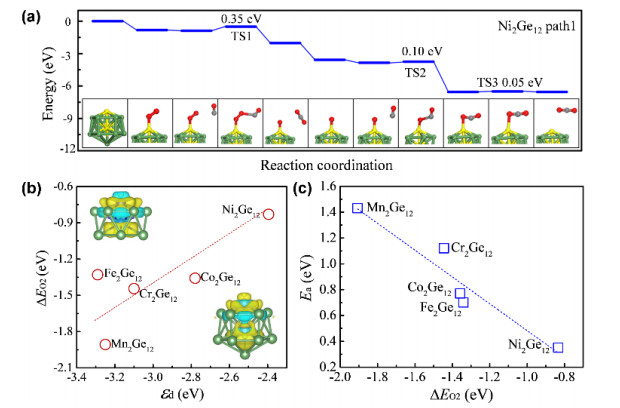
Not only singly doped, but also doubly doped cage clusters have been explored for catalytic applications. As mentioned above, the transition metal dual doped Ge12 clusters have been achieved in gas-phase experiment[19], in which one transition metal atom is located in the cage center, while the other one is exposed on the surface and thus can serve as a unique active site (see Fig. 2). By CGA-DFT search, we showed that the neutral Cr2Ge12 cluster is a Cr-centered hexagonal antiprism with a capped Cr atom (just like its anionic counterpart), and M2Ge12 (M = Mn, Fe, Co, and Ni) adopt an endohedral 13-vertex cage with an interior M atom[20]. As shown in Fig. 6, CO oxidation can occur on the surface metal atom of M2Ge12 under the Eley-Rideal mechanism. Ni2Ge12 exhibits the lowest reaction barrier of 0.35 eV, as well as good thermal stability and high resistance to agglomeration. The correlation between O2 binding strength (or reaction barrier) and the d orbital center of the two metal dopants was observed in these dual doped germanium clusters, suggesting universal regulation mechanism for subnanometer and bulk transition metal based catalysts.
Figure 6
Figure 6.
(a) Energy diagrams of CO oxidation on the Ni2Ge12 cluster, and the corresponding structures of elementary steps. Transition states and kinetic barriers are indicated. (b) Kinetic barrier of CO oxidation as a function of O2 adsorption energy. The C, O, Ge and Ni atoms are shown in grey, red, green and yellow, respectively. (c) Adsorption energy of O2 molecule on the M2Ge12 clusters as a function of the d orbital center (εd) of the cluster. The insets show the differential charge densities between the two metal dopants and Ge12 cage for Cr2Ge12 (upper panel) and Ni2Ge12 (lower panel) clusters, respectively. The yellow and cyan colors represent electron accumulation and depletion regions, respectively[20]
These theoretical explorations shine light in utilizing the experimentally accessible endohedral cage clusters for various catalytic reactions. Controllable size and geometry of clusters as well as doping element offer large degree of freedom to balance the activity and stability of these cage clusters. The established correlation between chemical activity and p or d orbital center would further help manipulate their catalytic performance with atomic precision.
5.
OUTLOOK
To end this perspective, we would like to point out several directions for future cluster research. Despite of the great success, the numerical efficiency of CGA-DFT scheme for globally searching PES is still limited, especially when the number of atoms approaches to 100. To further improve the robustness and efficiency, it would be desirable to take advantage of the emerging machine learning technique. For instance, a machine learning potential based on DFT database has been developed for boron clusters[44], which is 1~2 orders of magnitude faster than full DFT-based structure searches.
So far, the target of most structure prediction codes (including CGA) is the total energy. However, for a given cluster, some metastable isomers may exist at certain conditions. In order to design functional nanomaterials with desired properties, we will extend the capability of CGA code to inverse design by using one or a few key parameters of physical/chemical properties as the target function for global search. Together with the high-throughput exploration of many possible cluster sizes and compositions, one shall be able to truly design a functional cluster from scratch.
This perspective mainly focuses on our recent progress of doped clusters with endohedral cage structure. It is worthy to note that even an open-shell elemental cluster with core-shell geometry, e.g., Cu18−, could be highly stable and unreactive[45]. Deeper understanding of such cluster species would help design subnanometer clusters with precise catalytic behavior.
Finally, it would be demanding to add the functionality of CGA code to perform structure prediction and even inverse design on ligand-protected metal clusters and alloy clusters, as they were found to be promising catalysts for CO2 reduction and N2 fixation[46, 47].
[1]
Gong, X. G.; Kumar, V. Enhanced stability of magic clusters: a case study of icosahedral Al12X, X = B, Al, Ga, C, Si, Ge, Ti, As. Phys. Rev. Lett.1993, 70, 2078–2081. doi: 10.1103/PhysRevLett.70.2078
[2]
de Heer, W. A. The physics of simple metal clusters: experimental aspects and simple models. Rev. Mod. Phys.1993, 65, 611–676. doi: 10.1103/RevModPhys.65.611
[3]
Langmuir, I. Types of valence. Science, New Series1921, 54, 59–67.
[4]
Pyykkö, P.; Runeberg, N. Icosahedral WAu12: a predicted closed-shell species, stabilized by aurophilic attraction and relativity and in accord with the 18-electron rule. Angew. Chem.2002, 114, 2278–2280. doi: 10.1002/1521-3757(20020617)114:12<2278::AID-ANGE2278>3.0.CO;2-F
[5]
Li, X.; Kiran, B.; Li, J.; Zhai, H. J.; Wang, L. S. Experimental observation and confirmation of icosahedral W@Au12 and Mo@Au12 molecules. Angew. Chem. Int. Ed.2002, 41, 4786–4789. doi: 10.1002/anie.200290048
[6]
Heath, J. R.; O'Brien, S. C.; Zhang, Q.; Liu, Y.; Curl, R. F.; Kroto, H. W.; Tittel, F. K.; Smalley, R. E. Lanthanum complexes of spheroidal carbon shells. J. Am. Chem. Soc.1985, 107, 7779–7780. doi: 10.1021/ja00311a102
[7]
Kumar, V.; Kawazoe, Y. Metal-encapsulated fullerenelike and cubic caged clusters of silicon. Phys. Rev. Lett.2001, 87, 045503(1–4).
[8]
Kumar, V.; Kawazoe, Y. Metal-encapsulated caged clusters of germanium with large gaps and different growth behavior than silicon. Phys. Rev. Lett.2002, 88, 235504(1–4).
[9]
Koyasu, K.; Akutsu, M.; Mitsui, M.; Nakajima, A. Selective formation of MSi16 (M = Sc, Ti, and V). J. Am. Chem. Soc.2005, 127, 4998–4999. doi: 10.1021/ja045380t
[10]
Lau, J. T.; Hirsch, K.; Klar, P.; Langenberg, A.; Lofink, F.; Richter, R.; Rittmann, J.; Vogel, M.; Zamudio-Bayer, V.; Möller. T. X-ray spectroscopy reveals high symmetry and electronic shell structure of transition-metal-doped silicon clusters. Phys. Rev. A2009, 79, 053201(1–5).
[11]
Furuse, S.; Koyasu, K.; Atobe, J.; Nakajima, A. Experimental and theoretical characterization of MSi16−, MGe16−, MSn16−, and MPb16− (M = Ti, Zr, and Hf): the role of cage aromaticity. J. Chem. Phys.2008, 129, 064311(1–6).
[12]
Tsunoyama, H.; Akatsuka, H.; Shibuta, M.; Iwasa, T.; Mizuhata, Y.; Tokitoh, N.; Nakajima, A. Development of integrated dry-wet synthesis method for metal encapsulating silicon cage superatoms of M@Si16 (M = Ti and Ta). J. Phys. Chem. C2017, 121, 20507–20516. doi: 10.1021/acs.jpcc.7b06449
[13]
Zhao, J. J.; Du, Q. Y; Zhou, S.; Kumar, V. Endohedrally doped cage clusters. Chem. Rev. DOI: 10.1021/acs.chemrev.9b00651.
[14]
Huang, X. M.; Xu, H. G.; Lu, S. J.; Su, Y.; King, R. B.; Zhao, J. J.; Zheng, W. J. Discovery of a silicon-based ferrimagnetic wheel structure in VxSi12− (x = 1~3) clusters: photoelectron spectroscopy and density functional theory investigation. Nanoscale2014, 6, 4617–14621.
[15]
Huang, X. M.; Lu, S. J.; Liang, X. Q.; Su, Y.; Sai, L. W.; Zhang, Z. G.; Zhao, J. J.; Xu, H. G.; Zheng, W. J. Structures and electronic properties of V3Sin− (n = 3~14) clusters: a combined ab initio and experimental study. J. Phys. Chem. C2015, 119, 10987–10994. doi: 10.1021/jp5112845
[16]
Wu, X.; Zhou, S.; Huang, X. M.; Chen, M. D.; Bruce King, R.; Zhao, J. J. Revisit of large-gap Si16 clusters encapsulating group-Ⅳ metal atoms (Ti, Zr, Hf). J. Comput. Chem.2018, 39, 2268–2272. doi: 10.1002/jcc.25545
[17]
Zhou, S.; Yang, X. W.; Pei, W.; Zhao, J. J.; Du, A. J. Silicon nanocages for selective carbon dioxide conversion under visible light. J. Phys. Chem. C2019, 123, 9973–9980. doi: 10.1021/acs.jpcc.9b01784
[18]
Liang, X. Q.; Deng, X. J.; Lu, S. J.; Huang, X. M.; Zhao, J. J.; Xu, H. G.; Zheng, W. J.; Zeng, X. C. Probing structural, electronic, and magnetic properties of iron-doped semiconductor clusters Fe2Gen–/0 (n = 3~12) via joint photoelectron spectroscopy and density functional study. J. Phys. Chem. C2017, 121, 7037–7046. doi: 10.1021/acs.jpcc.7b00943
[19]
Liang, X. Q.; Kong, X. Y.; Lu, S. J.; Huang, Y. Y.; Zhao, J. J.; Xu, H. G.; Zheng, W. J.; Zeng, X. C. Structural evolution and magnetic properties of anionic clusters Cr2Gen (n = 3~14): photoelectron spectroscopy and density functional theory computation. J. Phys. : Condens. Matter2018, 30, 335501(1–11).
[20]
Zhou, S.; Yang, X. W.; Shen, Y. B.; King, R. B.; Zhao, J. J. Dual transition metal doped germanium clusters for catalysis of CO oxidation. J. Alloy. Compd.2019, 806, 698–704. doi: 10.1016/j.jallcom.2019.07.297
[21]
Du, Q. Y.; Wu, X.; Wang, P. J.; Wu, D.; Sai, L. W.; King, R. B.; Park, S. J.; Zhao, J. J. Structure evolution of transition metal-doped gold clusters M@Au12 (M = 3d~5d): across the periodic table. J. Phys. Chem. C2020, 124, 7449–7457. doi: 10.1021/acs.jpcc.9b11588
[22]
Zhou, S.; Pei, W.; Du, Q. Y.; Zhao, J. J. Foreign atom encapsulated Au12 golden cages for catalysis of CO oxidation. Phys. Chem. Chem. Phys.2019, 21, 10587–10593. doi: 10.1039/C9CP01517E
[23]
Zhao, J. J.; Shi, R. L.; Sai, L. W.; Huang, X. M.; Su, Y. Comprehensive genetic algorithm for ab initioglobal optimisation of clusters. Mol. Simulat.2016, 42, 809–819. doi: 10.1080/08927022.2015.1121386
[24]
Goldberg, D. E. Genetic algorithms in search. optimization, and machineLearning. Addison-Wesley1989.
[25]
Deaven, D. M.; Ho, K. M. Molecular geometry optimization with a genetic algorithm. Phys. Rev. Lett.1995, 75, 288–291. doi: 10.1103/PhysRevLett.75.288
[26]
Luo, Y. H.; Zhao, J. J.; Qiu, S. T.; Wang, G. H. Genetic-algorithm prediction of the magic-number structure of (C60)N clusters with a first-principles interaction potential. Phys. Rev. B1999, 59, 14903–14906. doi: 10.1103/PhysRevB.59.14903
[27]
Zhao, J. J.; Xie, R. H. Genetic algorithms for the geometry optimization of atomic and molecular clusters. J. Comput. Theor. Nanosci.2004, 1, 117–131. doi: 10.1166/jctn.2004.010
[28]
Wang, B. L.; Yin, S. Y.; Wang, G. H.; Buldum, A.; Zhao, J. J. Novel structures and properties of gold nanowires. Phys. Rev. Lett.2001, 86, 2046–2049. doi: 10.1103/PhysRevLett.86.2046
[29]
Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B1996, 54, 11169–11186. doi: 10.1103/PhysRevB.54.11169
[30]
Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys.2000, 113, 7756–7764. doi: 10.1063/1.1316015
[31]
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 1995, 117, 1–19. doi: 10.1006/jcph.1995.1039
[32]
Sai, L. W.; Wu, X.; Gao, N.; Zhao, J. J.; King, R. B. Boron clusters with 46, 48, and 50 atoms: competition among the core-shell, bilayer and quasi-planar structures. Nanoscale2017, 9, 13905–13909. doi: 10.1039/C7NR02399E
[33]
Huang, X. M.; Sai, L. W.; Jiang, X.; Zhao, J. J. Ground state structures, electronic and optical properties of medium-sized Nan+ (n = 9, 15, 21, 26, 31, 36, 41, 50 and 59) clusters from ab initio genetic algorithm. Eur. Phys. J. D2013, 67, 1–7. doi: 10.1140/epjd/e2012-30692-0
[34]
Sai, L. W.; Tang, L. L.; Huang, X. M.; Chen, G. B.; Zhao, J. J.; Wang, J. Lowest-energy structures of (WO3)n (2≤n≤12) clusters from first-principles global search. Chem. Phys. Lett.2012, 544, 7–12. doi: 10.1016/j.cplett.2012.06.050
[35]
Zhao, J. J.; Huang, X. M.; Jin, P.; Chen, Z. F. Magnetic properties of atomic clusters and endohedral metallofullerenes. Coordin. Chem. Rev.2015, 289–290, 315–340.
[36]
Kumar, V.; Kawazoe, Y. Metal-doped magic clusters of Si, Ge, and Sn: the finding of a magnetic superatom. Appl. Phys. Lett.2003, 83, 2677–2679. doi: 10.1063/1.1609661
[37]
Ma, L.; Zhao, J. J.; Wang, J. G.; Wang, B. L.; Lu, Q. L.; Wang, G. H. Growth behavior and magnetic properties of SinFe (n = 2~14) clusters. Phys. Rev. B2006, 73, 125439(1–8).
[38]
Kong, X. Y.; Xu, H. G.; Zheng, W. J. Structures and magnetic properties of CrSin− (n = 3~12) clusters: photoelectron spectroscopy and density functional calculations. J. Chem. Phys.2012, 137, 064307(1–9).
[39]
Wang, J. G.; Ma, L.; Zhao, J. J.; Wang, G. H. Structural growth sequences and electronic properties of manganese-doped germanium clusters: MnGen (2~15). J. Phys. : Condens. Matter2008, 20, 335223(1–8).
[40]
Claes, P.; Janssens, E.; Ngan, V. T.; Gruene, P.; Lyon, J. T.; Harding, D. J.; Fielicke, A.; Nguyen, M. T.; Lievens, P. Structural identification of caged vanadium doped silicon clusters. Phys. Rev. Lett.2011, 107, 173401(1–4).
[41]
Zhou, S.; Yang, X. W.; Xu, X.; Dou, S. X.; Du, Y.; Zhao, J. J. Boron nitride nanotubes for ammonia synthesis: activation by filling transition metals. J. Am. Chem. Soc.2020, 142, 308–317. doi: 10.1021/jacs.9b10588
[42]
Zhao, Y. Y.; Liu, N. S.; Zhou, S.; Zhao, J. J. Two-dimensional ZnO for the selective photoreduction of CO2. J. Mater. Chem. A2019, 7, 16294–16303. doi: 10.1039/C9TA04477A
[43]
Zhou, S.; Yang, X. W.; Pei, W.; Liu, N. S.; Zhao, J. J. Heterostructures of mxenes and N-doped graphene as highly active bifunctional electrocatalysts. Nanoscale2018, 10, 10876–10883. doi: 10.1039/C8NR01090K
[44]
Tong, Q. C.; Xue, L. T.; Lv, J.; Wang, Y. C.; Ma, Y. M. Accelerating CALYPSO structure prediction by data-driven learning of a potential energy surface. Faraday Discuss.2018, 211, 31–43. doi: 10.1039/C8FD00055G
[45]
Yin, B. Q.; Du, Q. Y.; Geng, L. J.; Zhang, H. Y.; Luo, Z. X.; Zhou, S.; Zhao, J. J. Anionic copper clusters reacting with NO: an open-shell superatom Cu18–. J. Phys. Chem. Lett.2020, DOI: 10.1021/acs.jpclett.0c01643.
[46]
Yang, D.; Pei, W.; Zhou, S.; Zhao, J. J.; Ding, W. P.; Zhu, Y. Controllable conversion of CO2 on non-metallic gold clusters. Angew. Chem. Int. Ed.2020, 59, 1919–1924. doi: 10.1002/anie.201913635
[47]
Sun, Y. N.; Pei, W.; Xie, M. C.; Xu, S.; Zhou, S.; Zhao, J. J.; Xiao, K.; Zhu, Y. Excitonic Au4Ru2(PPh3)2(SC2H4Ph)8 cluster for light-driven dinitrogen fixation. Chem. Sci.2020, 11, 2440–2447. doi: 10.1039/C9SC06424A
Figure 1
The lowest-energy structures of Fe2Gen (n = 3~12) clusters. The symmetries are given in parentheses. The red numbers (left and right) below each structure show magnetic moments (in unit of µB) for Fe1 and Fe2 atoms, respectively. Green and purple balls represent Ge and Fe atoms, respectively[18]
Figure 2
The lowest-energy structures of Cr2Gen− (n = 3~14) clusters. The red numbers (left and right) below each structure show magnetic moments (in unit of µB) for Cr1 and Cr2 atoms, respectively. Green and yellow balls represent Ge and Cr atoms, respectively[19]
Figure 3
Photoelectron spectra (upper) from experiment and theoretical calculation, and ground state structures (lower) of VxSi12− (x = 1, 2, 3) clusters[14]
Figure 4
(a) Structures of V@Sin (n = 12~16) clusters chemisorbed with a CO2 molecule, with C, O, Si, and V atoms represented in gray, red, yellow, and cyan colors, respectively. (b) The p orbital center (εp, black symbols) and coordination number (CN, red symbols) of Si atoms as a function of the size of V@Sin clusters. The insets display the differential charge density distributions between the V atom and Si cage. The electron accumulation and depletion regions are represented by red and green colors, respectively, using an isosurface value of 2 × 10−3 e/Å3. (c) Kinetic barrier (Ea) for chemisorption of CO2 and dissociation of the H2 molecule, and the barrier of the rate-limiting step for CO2 hydrogenation to various products on the V@Sin clusters[17]
Figure 5
(a) Reaction pathways of CO oxidation on the Cr@Au12 cluster under the L-H mechanism. The insets show the structures of the corresponding reaction intermediates and transition states (TS). The numbers indicate the kinetic barriers. The C, O, Cr and Au atoms are shown in grey, red, green and blue colors, respectively. (b) M−Au bond order and (c) d orbital center (εd) as a function of CO adsorption energy for various M@Au12 clusters. The insets in (b) show the differential charge density distributions of Cr@Au12 (Ih) and Ta@Au12 (Oh). The yellow and cyan colors represent the electron accumulation and depletation regions with isosurface value of 0.015 e/Å3, respectively[22]
Figure 6
(a) Energy diagrams of CO oxidation on the Ni2Ge12 cluster, and the corresponding structures of elementary steps. Transition states and kinetic barriers are indicated. (b) Kinetic barrier of CO oxidation as a function of O2 adsorption energy. The C, O, Ge and Ni atoms are shown in grey, red, green and yellow, respectively. (c) Adsorption energy of O2 molecule on the M2Ge12 clusters as a function of the d orbital center (εd) of the cluster. The insets show the differential charge densities between the two metal dopants and Ge12 cage for Cr2Ge12 (upper panel) and Ni2Ge12 (lower panel) clusters, respectively. The yellow and cyan colors represent electron accumulation and depletion regions, respectively[20]

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
