Login In
Login In
CoMFA Study on Anti-proliferative Activity of Fluoroquinolone Amide Derivatives
- Corresponding author: Jing-Pei CAO, caojingpei@cumt.edu.cn Chang-Jun FENG, fengcj@xzit.edu.cn
Figures(4)
Citation:
Hui FENG, Jing-Pei CAO, Chang-Jun FENG. CoMFA Study on Anti-proliferative Activity of Fluoroquinolone Amide Derivatives[J]. Chinese Journal of Structural Chemistry,
;2022, 41(3): 220324.
doi:
10.14102/j.cnki.0254-5861.2011-3343

Figures(4)





Gao, L. Z.; Xie, Y. S.; Li, T.; Huang, W. L.; Hu, G. Q. Synthesis, antiproliferative activity and SAR of C-3 oxadiazole sulfanyl-acetylhydrazone-substituted fluoroquinolone analogues. Acta Pharm. Sin. 2014, 49, 1694−1698.
Li, T.; Gao, L. Z.; Xie, Y. Y.; Feng, Y. F.; Yan, Q.; Wu, S. M.; Ni, L. L.; Zhao, H.; Huang, W. L.; Hu, G. Q. Synthesis and anti-proliferative activity of fluoroquinolone C-3 fused heterocyclic α, β-unsaturated ketones derived from ciprofloxacin. Acta Pharm. Sin. 2015, 50, 569−573.
Gao, L. Z.; Xie, Y. S.; Yan, Q.; Wu, S. M.; Ni, L. L.; Zhao, H.; Huang, W. L.; Hu, G. Q. Synthesis and anti-proliferative activity of fluoroquinolone(rhodanine unsaturated ketone) amide derivatives. Acta Pharm. Sin. 2015, 50, 1008−1012.
Abdulla, A. M.; Mei, Z.; Qiu, L.; Ju, X. H. Theoretical investigation on QSAR of (2-methyl-3-biphenylyl) methanol analogs as PD-L1 inhibitor. Chin. J. Chem. Phys. 2020, 33, 459–467.
doi: 10.1063/1674-0068/cjcp1909168
Zheng, S. S.; Li, T. T.; Wang, J.; Hu, Y. J.; Zhang, H. X.; Zhao, S. X.; Zhao, Y. H.; Li, C. QSAR models for predicting the aqueous reaction rate constants of aromatic compounds with hydrated electrons. Environ. Chem. 2019, 38, 1005–1013.
Ding, R.; Chen, J. W.; Yu, Y.; Lin, J.; Wang, Z. Y.; Tang, W. H.; Li, X. H. Using ensemble learning algorithms to develop QSAR models on bioconcentration factors of organic chemicals in multispecies fish. Environ. Chem. 2021, 40, 1295–1304.
Lei, B.; Zang, Y. L.; Xue, Z. W.; Ge, Y. Q.; Li, W.; Zhai, Q.; Jiao, L. Ensemble hologram quantitative structure activity relationship model of the chromatographic retention index of aldehydes and ketones. Chin. J. Chromatogr. 2021, 39, 331–337.
doi: 10.3724/SP.J.1123.2020.06011
Zheng, S. S.; Li, T. T.; Wang, J.; Hu, Y. J.; Zhang, H. X.; Zhao, S. X.; Zhao, Y. H.; Li, C. QSAR models for predicting the aqueous reaction rate constants of aromatic compounds with hydrated electrons. Environ. Chem. 2019, 38, 1005–1013.
Zeng, X. L.; Qu, R. J.; Feng, M. B.; Chen, J.; Wang, L. S.; Wang, Z. Y. Photodegradation of polyfluorinated dibenzo-p-dioxins (PFDDs) in organic solvents: experimental and theoretical studies. Environ. Sci. Tech. 2016, 50, 8128–8134.
doi: 10.1021/acs.est.6b02682
Qu, R. J.; Liu, J. Q.; Li, C. G.; Wang, L. S.; Wang, Z. Y.; Wu, J. C. Experimental and theoretical insights into the photochemical decomposition of environmentally persistent perfluorocarboxylic acids. Water Res. 2016, 104, 34–43.
doi: 10.1016/j.watres.2016.07.071
Feng, C. J. CoMFA model of inhibitory activity for nitrobenzene derivatives to photobacteria. J. Xuzhou Inst. Tech. (Nat. Sci. Ed. ) 2020, 35, 28–31.
Tong, J. B.; Qin, S. S.; Lei, S.; Wang, Y. A 3D-QSAR study of HIV-1 integrase inhibitors using RASMS and topomer CoMFA. Chin. J. Struct. Chem. 2019, 38, 867–881.
Shu, M.; Wu, T.; Wang, B. W.; Li, J.; Xu, C. M.; Lin, Z. H. 3D-QSAR and surflex docking studies of a series of alkaline phosphatase inhibitors. Chin. J. Struct. Chem. 2019, 38, 7–16.
Tong, J. B.; Zhan, P.; Bai, M.; Yao, T. Molecular modeling studies of human immunodeficiency virus type 1 protease inhibitors using three-dimensional quantitative structure-activity relationship, virtual screening, and docking simulations. J. Chemometr. 2016, 30, 523–536.
doi: 10.1002/cem.2809
Feng, C. J. CoMFA model of herbicidal activity of phenyl-sulfonylured derivatives. J. Xuzhou Inst. Tech. (Nat. Sci. Ed. ) 2019, 34, 21–25.
Feng, H.; Feng, C. J. CoMFA model of anti-proliferative activity for fluoroquinolon-3-yl s-triazole sulfide-ketone derivatives and implications for molecular design. Chin. J. Struct. Chem. 2021, 40, 703–710.
Feng, H.; Feng, C. J. 3D-QSAR studies on the anti-tumor activity of N-aryl-salicylamide derivatives. Chin. J. Struct. Chem. 2019, 38, 1874–1880.
Guo, Z. R. Molecular Drug Design. Science Press, Beijing 2005, p20.
Hu, S. Q.; Mi, S. Q.; Jia, X. L.; Guo, A. L.; Chen, S. H.; Zhang, J.; Liu, X. Y. 3D-QSAR study and molecular design of benzimidazole derivatives as corrosion inhibitors. Chem. J. Chin. Univ. 2011, 32, 2402–2409.
Cramer, R. D.; Burce, J. D.; Patterson, D. E.; Frank, I. E. Cross-validation, boot strapping, and partial least squares compared with multiple regression in conventional QSAR study. Quant. Struct. -Act. Relat. 1988, 7, 18–25.
doi: 10.1002/qsar.19880070105
Sheng Tang , Mingyue Liao , Weihai Sun , Jihuai Wu , Jiamin Lu , Yiming Xie . Optimizing CsPbBr3 perovskite solar cell interface and performance through tetraphenylethene derivatives. Chinese Chemical Letters, 2025, 36(6): 110838-. doi: 10.1016/j.cclet.2025.110838
Rongrong Zheng , Zuxiao Chen , Qiuyuan Li , Ni Yang , Wenjun Zhang , Chuyu Huang , Linping Zhao , Xin Chen , Hong Cheng , Shiying Li . Endoplasmic reticulum targeting photodynamic oxidizer to boost anti-tumor immunity by intensifying immunogenic cell death in conjunction with IDO1 inhibition. Chinese Chemical Letters, 2025, 36(12): 110865-. doi: 10.1016/j.cclet.2025.110865
Yang Liu , Jing Liang , Mengzhu Zheng , Haoze Song , Lixia Chen , Hua Li . PD-L1/SHP2 dual PROTACs inhibit melanoma by enhancing T-cell killing activity. Chinese Chemical Letters, 2025, 36(6): 110317-. doi: 10.1016/j.cclet.2024.110317
Wei Yan , Naizhen Zhang , Xiao Liu , Qiyu He , Xucheng Lv , Jianghui Sun , Lili Zhuang , Yuexin Zou , Yajie Zhang , Yuhang Liang , Yanjie Wang , Siyuan Li , Yonghui Sun . Development of visible-light photocaged molecular glues (vc-MGs) for B-cell malignancies therapy with improved safety and pharmacokinetic profiles in vivo. Chinese Chemical Letters, 2026, 37(5): 112064-. doi: 10.1016/j.cclet.2025.112064
Zixuan Chen , Yafeng Wu , Zhaoyan Tian , Zhaohan Wang , Weiwei Liu , Songqin Liu . A reproducible hybrid membrane for in situ analysis of cell secretions with a wide size range. Chinese Chemical Letters, 2025, 36(12): 110917-. doi: 10.1016/j.cclet.2025.110917
Xiaoyan Liu , Cong Xu , Ruhe Zhang , Yilu Zheng , Hengyu Liu , Haolin Chen , Meng Zhao , Jun Wu , Dongjun Lin . Black phosphorus nanosheets-based platform for B-cell lymphoma chemo-photothermal therapy. Chinese Chemical Letters, 2026, 37(5): 111401-. doi: 10.1016/j.cclet.2025.111401
Xiaopeng Han , Jiayin Li , Fei Li , Zhongyue Yuan , Hao Li , Lei Yang , Yan-Ming Xia , Chao Teng , Chao Qin , Lifang Yin . ROS-sensitive dihydroartemisinin prodrug amplify chemo-immunotherapy efficacy of doxorubicin by coordinating robust tumor cell immunogenic cell death and PD-L1 blockade. Chinese Chemical Letters, 2026, 37(2): 111335-. doi: 10.1016/j.cclet.2025.111335
Xiaoqin Pan , Changsheng Li , Xinyi Ai , Changqing Tao , Jingchuan He , Tingting Li , Zhihua Deng , Xiaocheng Mo , Ya Chen , Xiumei Qin , Dongmei Wang , Ronghua Jin , Jie Yang . Targeted delivery of nitidine chloride inducing ferroptosis and necroptosis as a novel anti-lung squamous cell carcinoma strategy. Chinese Chemical Letters, 2026, 37(6): 111968-. doi: 10.1016/j.cclet.2025.111968
Zhixue Liu , Haiqi Chen , Lijuan Guo , Xinyao Sun , Zhi-Yuan Zhang , Junyi Chen , Ming Dong , Chunju Li . Luminescent terphen[3]arene sulfate-activated FRET assemblies for cell imaging. Chinese Chemical Letters, 2024, 35(9): 109666-. doi: 10.1016/j.cclet.2024.109666
Yongjian Jiang , Feng Cheng , Jun Zhou , Lei Zhan , Chunmei Li , Chengzhi Huang . Regulation of cancer cell apoptosis with DNA nanocalculator. Chinese Chemical Letters, 2026, 37(1): 110071-. doi: 10.1016/j.cclet.2024.110071
Yunan Yuan , Zhimin Luo , Jie Chen , Chaoliang He , Kai Hao , Huayu Tian . Constructing thermoresponsive PNIPAM-based microcarriers for cell culture and enzyme-free cell harvesting. Chinese Chemical Letters, 2024, 35(7): 109549-. doi: 10.1016/j.cclet.2024.109549
Jisheng Liu , Junli Chen , Xifeng Zhang , Yin Wu , Xin Qi , Jie Wang , Xiang Gao . Red blood cell membrane-coated FLT3 inhibitor nanoparticles to enhance FLT3-ITD acute myeloid leukemia treatment. Chinese Chemical Letters, 2024, 35(9): 109779-. doi: 10.1016/j.cclet.2024.109779
Yulong Shi , Fenbei Chen , Mengyuan Wu , Xin Zhang , Runze Meng , Kun Wang , Yan Wang , Yuheng Mei , Qionglu Duan , Yinghong Li , Rongmei Gao , Yuhuan Li , Hongbin Deng , Jiandong Jiang , Yanxiang Wang , Danqing Song . Chemical construction and anti-HCoV-OC43 evaluation of novel 10,12-disubstituted aloperine derivatives as dual cofactor inhibitors of TMPRSS2 and SR-B1. Chinese Chemical Letters, 2024, 35(5): 108792-. doi: 10.1016/j.cclet.2023.108792
Cheng-Da Zhao , Huan Yao , Shi-Yao Li , Fangfang Du , Li-Li Wang , Liu-Pan Yang . Amide naphthotubes: Biomimetic macrocycles for selective molecular recognition. Chinese Chemical Letters, 2024, 35(4): 108879-. doi: 10.1016/j.cclet.2023.108879
Weiyu Chen , Zenghui Li , Chenguang Zhao , Lisha Zha , Junfeng Shi , Dan Yuan . Enzyme-modulate conformational changes in amphiphile peptide for selectively cell delivery. Chinese Chemical Letters, 2024, 35(12): 109628-. doi: 10.1016/j.cclet.2024.109628
Zeyang Yao , Xinru You , Xudong Wang , Yunze Kang , Liying Wang , Ziji Zhang . Stem cell-based hydrogel for the repair and regeneration of cartilage. Chinese Chemical Letters, 2025, 36(8): 110607-. doi: 10.1016/j.cclet.2024.110607
Yuanyi Zhou , Lili Wang , Li Chen , Qingbing Zha , Yu Meng , Mingshan Zhu . Functional inorganic nanomaterials for renal cell carcinoma treatment: Advancements and trends. Chinese Chemical Letters, 2025, 36(12): 110994-. doi: 10.1016/j.cclet.2025.110994
Huihui Wei , Yiming Niu , Mingli Xie , Weining He , Min Chen , Tenghui Wang , Qinhong Jiang , Panfei Xing , Chunming Wang . Photo-bioactivated azido fluorophores for passive and active cell imaging. Chinese Chemical Letters, 2026, 37(6): 111409-. doi: 10.1016/j.cclet.2025.111409
Juan He , Jiao-Xian Du , Meng Wang , Xiao-Dong Luo , Tao Feng . Irpexlactones A and B, a pair of ring-rearranged tremulane sesquiterpenoids from the basidiomycete Irpex lacteus and their anti-inflammatory activity. Chinese Chemical Letters, 2025, 36(10): 110769-. doi: 10.1016/j.cclet.2024.110769
Kun-Heng Li , Hong-Yang Zhao , Dan-Dan Wang , Ming-Hui Qi , Zi-Jian Xu , Jia-Mi Li , Zhi-Li Zhang , Shi-Wen Huang . Mitochondria-targeted nano-AIEgens as a powerful inducer for evoking immunogenic cell death. Chinese Chemical Letters, 2024, 35(5): 108882-. doi: 10.1016/j.cclet.2023.108882
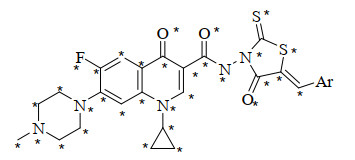
Ar: 1* = ph, 2 = 4-MeOph, 3 = 2-MeOph, 4 = 3, 4-(OCH2O)ph, 5* = 3, 4, 5-(MeO)3ph, 6 = 4-Meph, 7 = 4-Clph, 8 = 4-Fph, 9 = 4-NO2ph, 10 = 3-NO2ph, 11* = pyridine-4-yl, 12 = pyridine-3-yl, 13 = pyridine-2-yl, 14 = furan-2-yl, 15 = 4-HO2Cph, 16 = 3-HO2Cph, 17* = 4-NCph, 18 = 4-H2NSO2ph, 19* = 2-H2NSO2ph, 20* = 2, 3, 4-(H2NSO2)3ph, 21* = 2, 4-(HO2C)2ph, 22* = 2, 3, 4-(HO2C)3ph Note: (1) "*" represents the compounds in the test set. (2) 19~22 are the newly designed molecules.
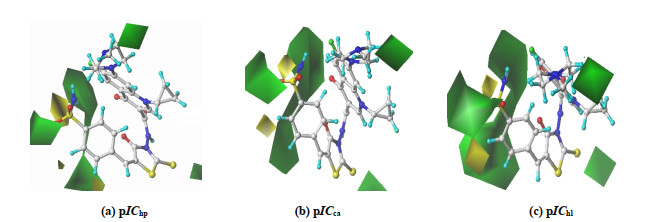
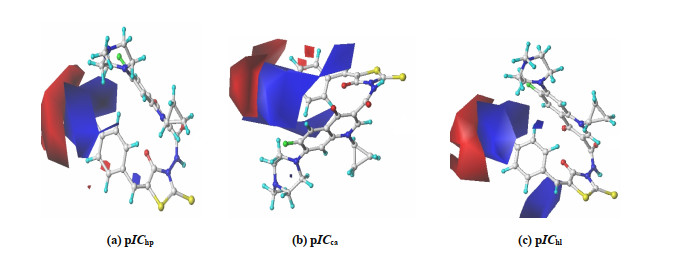
(a) pIChp (b) pICca (c) pIChl
 DownLoad:
DownLoad: