
 Login In
Login InHighly N2 dissociation catalyst: Ir(100) and Ir(110) surfaces
-
* Corresponding authors.
E-mail addresses: zhaochenxu@xatu.edu.cn (C. Zhao), ful263@nenu.edu.cn (L. Fu).
Figures(5)
Citation:
Chaozheng He, Menghui Xi, Chenxu Zhao, Ran Wang, Ling Fu, Jinrong Huo. Highly N2 dissociation catalyst: Ir(100) and Ir(110) surfaces[J]. Chinese Chemical Letters,
;2025, 36(3): 109671.
doi:
10.1016/j.cclet.2024.109671

Figures(5)




Due to the rapid growth of the world economy, the emergence of problems such as energy shortage and severe environmental pollution are threatening the survival of mankind, so there is an urgent need to develop a clean, renewable and environmentally friendly way to develop energy sources [1-4]. Ammonia is a very important chemical product in industrial production, which is widely used in the chemical industry, fertilizers, synthetic fibers and other fields [5-9]. In addition, ammonia is also considered to be a highly promising alternative energy fuel to hydrogen, not only due to its high hydrogen content density (17.6 wt%), but also because it is a zero-carbon energy storage product [10-12]. In particular, ammonia is more easily stored and transported as liquid fuel compared to gaseous hydrogen [13-15]. Nitrogen reduction reaction (NRR) is a promising way for ammonia synthesis and electrochemical method has attracted a lot of attention due to its clean and sustainable characters [16-19]. Many pioneering studies have successfully designed NRR catalysts with high reactivity and selectivity. Zheng et al. have predicted Ni3NG, Ru3NG, and Ir3NG as promising electrocatalysts for NRR with very low limiting potentials of −0.31 V to −0.33 V [20]. Moreover, Meng et al. identified W@MoSSe and Os@MoSSe as effective candidate catalysts for NRR on the basis of estimated stability, high catalytic activity and unchallengeable selectivity, where W@MoSSe was screened as the best NRR catalyst with a limiting potential of −0.15 V [21]. Overall, high performance of N2 activation and ammonia synthesis has been achieved in pioneering works. Moreover, the mechanisms of NRR have also been comprehensively summarized, including remote, alternating, and enzymatic mechanisms, on various kinds of catalysts. However, there are currently very few NRR catalysts that can dissociate N2 directly (denoted as dissociation mechanism). This is because that the cleavage of the N≡N triple bond requires an ultrahigh energy consumption of 410 kJ/mol [22-25]. The prerequisite of the dissociation mechanism is the strong interaction between N2 and the catalyst, which can lead to effective N2 activation. However, the direct dissociation of N2 is still challenged, even if N2 is successfully activated. Therefore, it is urgently needed to search for high performance catalysts for N2 dissociation [5,10,20,26].
Noble metal catalysts, such as Pd, Ru, Ir, always possess high reactivity for activation of inert gases due to the unoccupied d-orbitals [7,19,27,28]. In addition, these catalysts have excellent binding properties and electrical conductivity, resulting in excellent catalytic performance and are widely used in electrochemical reactions. Taking Ru as an example, Du et al. [29] have performed NRR on Ru(0001) with V doped on it. The results have shown a low negative limiting potential of −0.15 V for NRR to NH3. Mainly, the alloying of Ru with transition metals can modulate the binding strength of NRR intermediates and eventually improve its catalytic activity. Au as one of the most effective catalysts for NRR is also a noble metal that has been widely studied due to its high affinity and ability to activate N2 for electrochemical reduction as well as its generally low tendency of hydrogen formation [30]. Among numerous noble metals, Ir is also a promising catalyst, which has been widely used in recent years. Many pioneering works have focused on improving the utilization rate of Ir atom, which can effectively realize high reactivity [31-34]. For example, Strickler et al. [35] have performed DFT calculations on bimetallic Ir-based films and demonstrated the tendency between activity and stability of oxygen evolution reaction (OER) at 1.65 V vs. RHE. in acidic media. He et al. [34,36] have systematically studied the adsorption and dissociation of NO on Ir(100) for the formation of N2. The results have shown that the dissociation of NO into N and O only needs to overcome an energy of 0.46 eV. The dissociated N atoms can also combine into N2 with high selectivity. Song et al. [37,38] have performed NRR on Ir(100) doped with 3d transition metal atoms. Through regulation of electronic structure, an ultralow limiting potential of −0.36 V has been achieved, making it a potential NRR electrocatalyst. Although many studies on Ir have been successfully performed, the mechanism of N2 activation and dissociation on Ir still remains elusive.
Inspired by the pioneering studies, we herein study the dissociation mechanism of N2 molecules on Ir(100) and Ir(110) and analyze the relevant properties of N2 activation based on density functional theory (DFT). Through the analysis of adsorption energy, reaction barrier and optimal adsorption site, the hollow (H) site was finally determined to be a favorable dissociation site for N2. The N2 dissociation should overcome an energy barrier of 1.12 eV on Ir(110), while it is only 0.87 eV for Ir(100). Thus, Ir(100) was screened as a promising catalyst for N2 dissociation compared to Ir(110). This can be attributed to the fact that the adsorption energy of N2 at the H site of Ir(100) (−0.48 eV) is significantly higher than that of Ir(110) (−0.22 eV), resulting in different dissociation mechanisms for Ir(100) and Ir(110). Our results have not only pointed out the essential reason for choosing the NRR reaction on the crystal surface of Ir(100), but also provide a good idea for the laboratory workers to choose the precious metal Ir catalyst with high activity.
All the calculations were performed in Vienna Ab into Simulation Package (VASP) by using spin-polarized density functional theory (DFT) [39-42]. The electronic exchange-correlation interactions were described by the Perdew and Wang (PW91) functional within generalized gradient approximation (GGA) [43,44]. The electron-ion interactions were described by the projector augmented wave (PAW) method [45-49]. A cutoff energy of 400 eV was adopted for the planewave basis set. The convergence criteria for energy and force were set to be 10−5 eV and 0.05 eV/Å, respectively. The parameters of Ir unit-cell were set according to the experimental results (3.88 Å vs. 3.84 Å) [50]. The 2 × 2 × 1 supercell of the Ir(100) and Ir(110) facet with five layers was taken as the substrate model. Brillouin zones were sampled by a Monkhorst-Pack k-point mesh with a 7 × 7 × 1 k-points grid for Ir(100) and 3 × 5 × 1 k-points grid for Ir(110). To avoid the interaction between two neighboring surfaces, a vacuum space over 20 Å was used. The bottom two atomic layers were fixed in their bulk positions whereas the remaining top layers and the adsorbates were allowed to relax. In the calculations of gas adsorption, 15 Å vacuum layer was used to avoid interactions between the neighboring images. Considering the interaction between layers, a layer spacing of over 15 Å is sufficient to overcome the interaction between layers. The climbing nudged elastic band(CI-NEB) method was used to search the saddle points and minimum energy pathways [51]. The transition states were also verified by its one imaginary vibrational frequency [52].
Adsorption energy is defined as the change of system energy before and after adsorption, which can be calculated as follows:
|
|
(1) |
where Etot denotes the total energy of the N2-supported Ir atom. Esub and EN denote the optimized atomic surface and the energy of isolated nitrogen molecules, respectively.
We start our study from the characters of catalyst configurations. To investigate the N2 dissociation, we have constructed different Ir surfaces, including Ir(100) and Ir(110), and the optimized structures are shown in Figs. S1a and b (Supporting information). The Ir(100) and Ir(110) exhibit different characters: The Ir(100) surface is flat compared to Ir(110), which has some ridges and grooves on the surface. The different characters can directly lead to various bond length of Ir-Ir, which is 2.714 Å on Ir(100) and 2.694 Å on Ir(110). These results are in agreement with the pioneering studies [34].
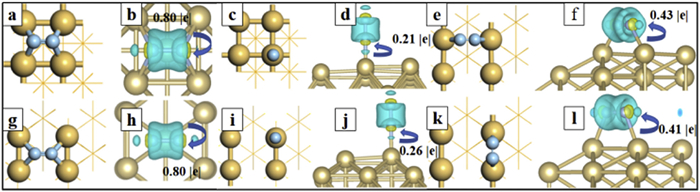
In order to confirm the adsorption behavior of N2, we have considered many adsorption sites, including bridge (B), top (T) and hollow (H) positions, on Ir(100) and Ir(110), as labeled in Fig. S1 (Supporting information). In addition, the bridge positions can be divided into long bridge (LB) sites and short bridge (SB) sites. After calculation of adsorption energies, the optimal adsorption site are confirmed as T site (LB site) on Ir(100) (Ir(110)) with an adsorption energy of −0.87 eV (−1.12 eV). The adsorption strength has indicated that there is a strong interaction between N2 molecules and substrates via chemisorption, which is beneficial for the activation of N2 molecules. This can also be reflected from the distance between Ir surface and adsorbed N2 (the dN-Ir in Table S1 in Supporting information). The bond lengths of N—Ir (dN-Ir) bonds are generally less than 2.0 Å, which is much smaller than the sum of the radii of the N and Ir atoms (2.63 Å). In order to quantitatively reveal the interaction between N2 and Ir, we focus on the bond lengths of N≡N (dNN) of N2 adsorbed on Ir. Compared to the N2 molecule in the free state, the N≡N bond lengths of the adsorbed N2 on Ir are all somewhat elongated. Among them, the N2 adsorbed on the H site of Ir(100) via horizontal manner can be activated most effectively with the longest N≡N bond of 1.294 Å, which has significantly elongated compared to the situation in gas phase (1.12 Å). This phenomenon can be interpreted from the coordination environment of active sites: N2 binds with Ir with only one Ir atom at the T-site and four Ir atoms at the H-site. The four-atom-coordination interaction can lead to more charge transfer between Ir and N2, which is assumed to be a beneficial factor for N2 activation. In fact, the calculated charge transfer (QCT) is consistence with our consumption: The QCT is increased with increasing coordination number between N2 and Ir, which can be described as QCT[T] (−0.26 e) < QCT[SB] (−0.41 e) < QCT[LB] (−0.43 e) < QCT[H] (−0.8 e), where the QCT[T] (QCT[SB], QCT[LB] QCT[H]) denotes the charge transfer between Ir and N2 adsorbed on the T (SB, LB, H) site.
To reveal the intrinsic mechanism of N2 activation on Ir, we have performed some calculations on electronic properties, including charge density difference (CDD), density of states (DOS), and crystal orbital Hamiltonian group (COHP).
For CDD analysis, we have considered the charge distribution between Ir and N2, adsorbed on various positions (Fig. 1). The cyan part in CDD denotes the depletion of electrons and the yellow part denotes the accumulation of electrons. Obviously, a significant charge transfer is observed between Ir and N2, denoting effective activation of adsorbed N2. Clearly, the charge density distribution around the N2 molecule is cylindrical, the charge distribution outside the column shows depleted trend and it is accumulated inside the column. In addition, both charge accumulation and reduction can be observed around the adsorbed N2 and the Ir atom connected to it. These characters perfectly accord with the “acceptance-donation” mechanism proposed in pioneering works. Ir manifests the valence electrons of 5d76s2 and the Ir-d orbitals are half occupied. The empty Ir-d orbitals can accept lone-pair electrons of N2 and the separated Ir-d electrons can also be donated into the anti-binding orbitals of N2, which can effectively weaken the N≡N triple bonds and facilitate the N2 dissociation [39]. Overall, the N2 is negatively charged on both Ir(100) and Ir(110), which is in accordance with the bader charge analysis (Table S1). Due to the larger electronegativity of N compared to Ir atoms, the charge will transfer from Ir to N2 with a large amount of −0.79 e (−0.80 e) for Ir(100) (Ir(110)), which is beneficial for the dissociation of N≡N bond. The figures in Fig. 1 are all periodic in the planar directions and it is vacuum in the vertical directions. This is a common slab model to simulate the surface of bulk metals.
To further reveal the potential activation mechanism of N2 molecules, we have calculated the projected crystal orbital Hamiltonian group (COHP) and partial density of states (PDOS) to reveal the bond properties between N2 and Ir from the aspect of electronic configuration (Fig. 2). COHP analysis is widely used to characterize bonding character with positive and negative values denoting bonding and anti-bonding contribution, respectively.
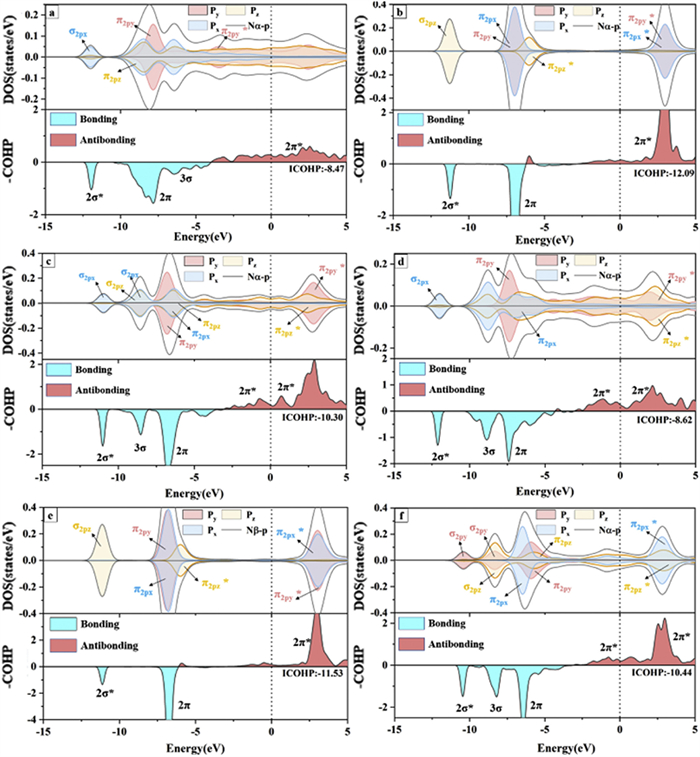
From the COHP analysis, we can see that the bonding states for the adsorbed N≡N bonds mainly contributed by the 2σ*, 2π and 3σ orbitals, corresponding to the σ2s*, σ2px, π2py and π2pz orbitals, respectively. In addition, the adsorbed N2 is negatively charged with −0.21 ~ −0.80 e and large amounts of 2π* states of the adsorbed N2 locate below the Fermi level. This can indicate that the electrons from Ir have dramatically filled the 2π* orbitals of N2 as N2 adsorbed on Ir. These phenomena illustrate that the bonding level of the N≡N triple bond is weakened and N2 has been activated effectively. The bond length of N2 adsorbed on the hollow/top site of Ir(100) has elongated to 1.294/1.127 Å compared to 1.10 Å in the free state. More interestingly, the 2π, 2π* and 3σ orbitals of N2 adsorbed at the hollow site of Ir(100) undergo a significant energy level splitting compared to the top site, indicating an increased spin density. The unpaired electrons induced by the spin state are beneficial for the protonation of N2. We have also integrated the crystal orbital Hamilton population (ICOHP) up to Fermi level, to quantitatively depict the N2 activation. The ICOHP values can reflect how much this bond contributes to the band-structure energy. ICOHP refers to the numerical integral from the lowest energy peak to 0 eV on the COHP. In general, the bonding (anti-bonding) state is defined as negative (positive) in COHP. Therefore, ICOHP can quantitatively indicate the occupation degree of anti-bonding states: The less negative ICOHP of N2 denotes a higher occupation of antibonding orbitals, corresponding to a more activated degree of N2. We have measured the activation degree of N2 from the aspect of N—N bond length in the above discussions. However, the change of N—N bond length is not the only factor to decide the activation ability. We have therefore further demonstrated the activation of N2 quantitatively from ICOHP. The ICOHP of N≡N bond for N2 adsorbed at hollow site (−8.47) is significantly less negative than the situation for top site (−12.07), which further indicates that the hollow site is more favorable for N2 activation compared to the top site. Similar conclusions can also be obtained for other sites: The ICOHP of N≡N bonds for N2 adsorbed on LB (−10.30) and SB (−10.44) sites all possess more negative value than that for H site. From the partial density of states, we can also see that the filled 2π* orbitals of N2 adsorbed on Ir(110) (Ir(100)) are mainly contributed by the states of N-π2py* and N-π2pz* (N-π2py*). Shortly, the Ir atoms can contribute large amount of electrons to the p-orbitals of N2, leading to the full activation of N2 at the hollow position of the Ir(100) crystal plane. In addition, the vacancy position of the Ir(100) crystal plane with an ICOHP value of −8.47 eV is more conducive to the activation of N2 than the hollow position of Ir(110) with an ICOHP value of −8.62 eV. To further demonstrate the correlation between ICOHP and N—N bond length, we eventually comprehensively compare the numeric values on various active sites. For Ir(100), the N—N bond length of adsorbed N2 is 1.13 Å (1.29 Å) on T (H) site, corresponding to a N—N ICOHP of −12.09 (−8.47). For Ir(110), the N—N bond length of N2, adsorbed on T, (B and H) site, is 1.13 Å (~1.19 Å and 1.30 Å), with an N—N ICOHP of −11.53 (~−10.40 and −8.62). Therefore, a more enlonged N—N bond corresponds to a higher occupation degree of antibonding states, which is beneficial for N2 activation. Therefore, the change of N—N bond length is eventually proved as a valid criterion to decide the activation ability of catalysts for N2.
These phenomena can be treated as the origin of elongated N≡N bond length and shortened N—Ir bond (Table S1). Overall, the hollow site can be treated as the most favorable adsorption sites for N2 activation compared to other sites.
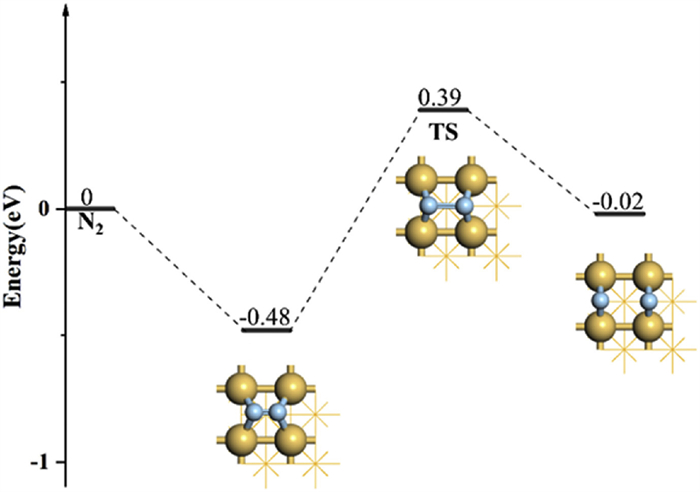
To explore the activation ability of N2 on Ir quantitatively, we have calculated the reaction pathway of N2 dissociation, including the initial states, transition states (TS), and final states (Fig. 3). The adsorption free energy of N2 on the hollow site of Ir(100) is −0.48 eV and the reaction barrier of N2 dissociation is 0.87 eV, which can be easily overcome at ~50 ℃. The N≡N bond length of adsorbed N2 has been elongated to 1.94 Å compared to 1.29 Å for the initial state.
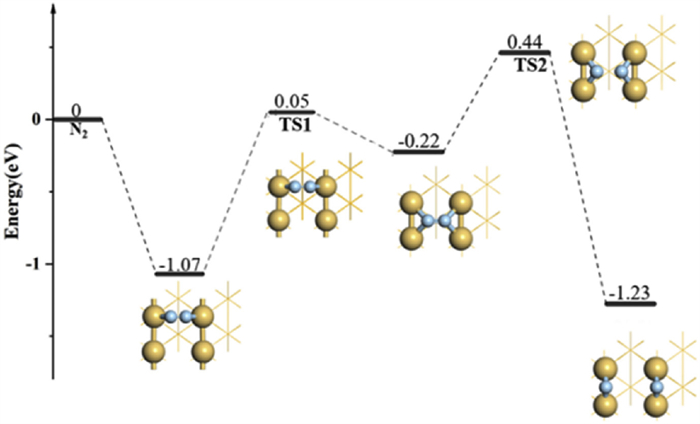
We have also considered the dissociation of N2 molecules on Ir(110) (Fig. 4). Unlike the situation on Ir(100), the adsorption energy of N2 is only −0.22 eV on the H site, indicating an ultralow capture property of N2. Therefore, the N2 prefers to be adsorbed at LB site, with an adsorption energy of −1.07 eV, and further experiences a movement from LB to H site with a barrier of 1.12 eV The transition state (TS1) has an imaginary frequency of −199.95 cm−1 and the corresponding N≡N bond of adsorbed N2 has extended from 1.179 Å (initial state) to 1.231 Å (final state). The following step is the dissociation of N2 at the hollow site with a reaction barrier of 0.66 eV. The N≡N bond length of adsorbed N2 in transition state (TS2) is further elongated to 1.826 Å with a virtual frequency of 459.7 cm−1. Finally, the N2 has successfully dissociated into two N atoms in the last step. This can reflect a synergistic effect between LB and H sites: The LB (H) site is superior in the capture (activation) ability of N2, compared to the situation of H (LB) site. There indeed exists some correlations between the capture and activation capacities of molecules: An increased activation property can lead to a decreased capture ability. This can be treated as a common rule for the activation of small molecules, such as N2, CO2, and O2. In our previous work, we have also discussed the activation of CO2 on Pt atom loaded on substrates, including BC3N2 and graphene. The results have shown that the N2 adsorbed on BC3N2 system manifests a higher occupation degree of antibonding orbitals, corresponding to a higher activation property. However, the BC3N2 system cannot effectively capture N2 due to the dramatic filled antibonding states, compared to the situation of graphene system [53].
Finally, we comprehensively compared the situations on Ir(100) and Ir(110). N2 molecule can be dissociated with an energy barrier of 0.87 eV (1.12 eV) on Ir(100) (Ir(110)). Based on rough rule-of-thumb, the energy barrier can be overcome at about 348 K (448 K) for Ir(100) (Ir(110)). Clearly, the temperature required for N2 dissociation on Ir(100) is significantly lower compared to the situation on Ir(110). This can be interpreted from the different reaction mechanism between Ir(100) and Ir(110): The N2 can be directly dissociated into N atoms on Ir(100) surface; In comparison, the dissociation of N2 on Ir(110) need to experience two steps, including migration and dissociation processes (Fig. 4), with two transition states. This may originate from the different capture ability of H site between Ir(100) and Ir(110). The adsorption energy of N2 is −0.48 eV on H site of Ir(100), indicating a favorable capture ability for N2. Therefore, the N2 prefers to be directly adsorbed and dissociated on the hollow site of Ir(100). In comparison, the adsorption energy of N2 is only −0.22 eV on the hollow site of Ir(110), which is unfavorable for N2 capture. Therefore, N2 prefers to be adsorbed on the bridge site of Ir(110) and further transfers to the hollow site. Finally, the N2 will dissociate into separated N atoms on the hollow site. About the adsorption process of N2, the N2 in free state can adsorb on the surfaces of Ir(100) and Ir(110) with an energy release of 0.48 eV and 1.07 eV, respectively. Therefore, there is no transition state for the process of N2 adsorption (Fig. S4 in Supporting information).
To better demonstrate that the most critical factor of ammonia synthesis is N2 dissociation, we have calculated the entire reaction pathway of N2 electroreduction to NH3. It can be seen from Fig. 5 that once N2 has dissociated, the subsequent hydrogenation steps can generally proceed spontaneously with negative reaction energies. Thus, the N2 dissociation process can be treated as the rate controlling step of ammonia synthesis, which is accordance with the key point of our research. As mentioned above, the barrier of N2 dissociation can be overcome at 348 K. We therefore performed the corresponding correction of free energy with temperature of 348 K, and the reaction pathway is displayed in Figs. 5c and d. The protonation steps of dissociated N atoms have also demonstrated negative reaction energies, similar to the situation at room temperature. Therefore, we can assume that the N2 dissociation and the following ammonia synthesis processes can occur spontaneously once the temperature higher than 348 K on Ir(100). In addition to the reaction pathway, we can also demonstrate the factors for ammonia synthesis from the aspect of charge transfer. The Ir atom has unpaired electrons, which can accept lone-pair electrons from N2. This can reflect that the N≡N bond can be weakened via the well-known “accept-donate” mechanism. Therefore, we can also attempt to increase the charge density of Ir to promote the occupation of antibonding orbitals to activate N2.
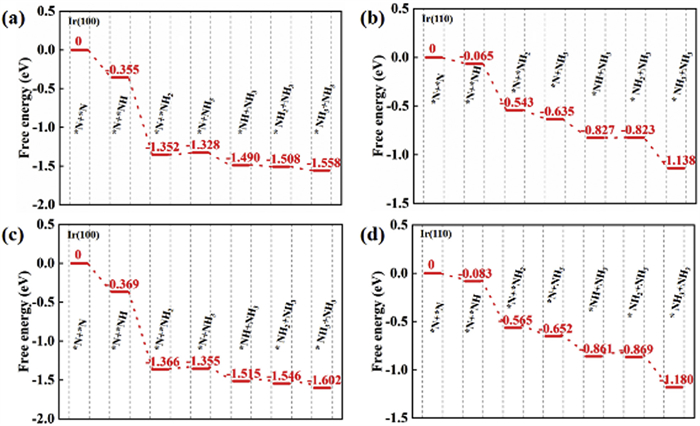
Considering the realistic condition, some ambient adsorbate such as *N2 may induce catalysts’ dynamics or reconstructions. We thus adsorb *N2 on Ir(100) (denoted as N2/Ir(100)) and test the stability (Fig. S2 in Supporting information). The N2/Ir (100) structure has shown negligible deformation after 10 ps dynamic calculation at 300 K. In addition, the structural integrity was maintained with no bond breaking, indicating a strong chemical bonds character between N2 molecule and Ir (100). Moreover, the energy of the system is nearly constant throughout the dynamic simulation, further indicating the good stability of N2/Ir (100) complex. In addition, in the real electrochemical condition, the high-index surfaces of Ir can also anticipate in the reaction. We have therefore calculated the free energy profile of N2 dissociation on Ir(211) surface to demonstrate the superiority of Ir(100) and Ir(110). As shown in Fig. S3 (Supporting information), the dissociation barrier of N2 on Ir(211) is 2.63 eV, which is significantly higher than that on Ir(100) and Ir(110) (0.87 eV and 1.12 eV). Therefore, the low-index surfaces of Ir can be treated as the main active centers for N2 dissociation in the realistic condition.
In conclusion, in this paper, the adsorption and dissociation of N2 on Ir(100) and Ir(110) were investigated using density functional theory. The results can be listed as follows: Ir(100) can interact with N2 effectively with an adsorption energy of −0.48 eV, leading to an ultralow N2 dissociation-barrier of 0.87 eV; In contrast, the Ir(110) can effectively capture N2 with a large adsorption energy of −1.07 eV, but the dissociation of N2 is a little difficult with a barrier of 1.12 eV. Therefore, the Ir(100) can be treated as a promising catalyst for N2 dissociation compared to Ir(110). This can be attributed to the significantly higher adsorption energy of N2 on the H site of Ir(100) (−0.48 eV) compared to the H site of Ir(110) (−0.22 eV), leading to different dissociation mechanisms on Ir(100) and Ir(110). Ir(100) can dissociate N2 directly on H site and Ir(110) should firstly capture N2 via LB site and further transfer the adsorbed N2 to the H site, which will dramatically deteriorate the reactivity of N2 dissociation. In addition, the barrier of N2 dissociation can be easily overcome at a low temperature of 348 K, at which the reaction energies of the following protonation steps of dissociated *N atoms are all negative. Therefore, as the temperature higher than 348 K, the N2 dissociation and ammonia synthesis can occur spontaneously on Ir(100) surface. Our calculations have provided clues for further catalyst design of N2 dissociation and ammonia synthesis in both theory and experiments.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This study was funded by the Natural Science Foundation of China (No. 21603109), the Henan Joint Fund of the National Natural Science Foundation of China (No. U1404216), the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0676). This work was also supported by Natural Science Basic Research Program of Shanxi (Nos. 2022JQ-108, 2022JQ-096).
Supplementary material associated with this article can be found, in the online version, at doi:
L. Fu, R. Wang, C.X. Zhao, et al., Chem. Eng. J. 414 (2021) 128857.
doi: 10.1016/j.cej.2021.128857
J. Huo, L. Fu, C. Zhao, et al., Chin. Chem. Lett. 32 (2021) 2269–2273.
doi: 10.1016/j.cclet.2020.12.059
R. Wang, C. He, W. Chen, et al., Chin. Chem. Lett. 32 (2021) 3821–3824.
doi: 10.1016/j.cclet.2021.05.024
J. Li, S. Chen, F. Quan, et al., Chem 6 (2020) 885–901.
doi: 10.3390/polym12040885
V. Rosca, M. Duca, M.T. de Groot, et al., Chem. Rev. 109 (2009) 2209–2244.
doi: 10.1021/cr8003696
S. Licht, B. Cui, B. Wang, et al., Science 345 (2014) 637–640.
doi: 10.1126/science.1254234
X. Lu, J. Zhang, W.K. Chen, et al., Nanoscale Adv. 3 (2021) 1624–1632.
doi: 10.1039/d1na00015b
Z. Huang, M. Rafiq, A.R. Woldu, et al., Coord. Chem. Rev. 478 (2023) 214981.
doi: 10.1016/j.ccr.2022.214981
Y. Ying, K. Fan, X. Luo, et al., Mater. Adv. 1 (2020) 1285–1292.
doi: 10.1039/d0ma00348d
M. Bat-Erdene, G. Xu, M. Batmunkh, et al., J. Mater. Chem. A 8 (2020) 4735–4739.
doi: 10.1039/c9ta13485a
T. Lan, Y. Zhao, J. Deng, et al., Catal. Sci. Technol. 10 (2020) 5792–5810.
doi: 10.1039/d0cy01137a
C. He, J. Wang, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 1051–1057.
doi: 10.1016/j.cclet.2021.09.009
Z. Chen, J. Zhao, C.R. Cabrera, et al., Small Methods 3 (2018) 1800368.
Z. Chen, J. Zhao, L. Yin, et al., J. Mater. Chem. A 7 (2019) 13284–13292.
doi: 10.1039/c9ta01410a
Z.M. Zhang, X. Yao, X.Y. Lang, et al., Appl. Surf. Sci. 536 (2021) 147706.
doi: 10.1016/j.apsusc.2020.147706
C. He, H. Wang, L. Fu, et al., Chin. Chem. Lett. 33(2022) 990–994.
doi: 10.1016/j.cclet.2021.09.049
X. Lv, W. Wei, B. Huang, et al., Nano Lett. 21 (2021) 1871–1878.
doi: 10.1021/acs.nanolett.0c05080
D. Ma, Y. Wang, L. Liu, et al., Phys. Chem. Chem. Phys. 23 (2021) 4018–4029.
doi: 10.1039/d0cp04843g
H. Shen, C. Choi, J. Masa, et al., Chem 7 (2021) 1708–1754.
doi: 10.1016/j.chempr.2021.01.009
X. Zheng, Y. Liu, Y. Yao, Chem. Eng. J. 426 (2021) 130745.
doi: 10.1016/j.cej.2021.130745
Y. Meng, T. Wang, J. Chen, et al., Appl. Surf. Sci. 640 (2023) 158470.
doi: 10.1016/j.apsusc.2023.158470
X. Liu, Y. Geng, R. Hao, et al., Progr. Chem. 33 (2021) 1074–1091.
L. Niu, L. An, X. Wang, et al., J. Energy Chem. 61 (2021) 304–318.
doi: 10.1016/j.jechem.2021.01.018
C. He, R. Sun, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 527–532.
doi: 10.1016/j.cclet.2021.05.072
L. Lin, L. Yan, L. Fu, et al., Fuel 308 (2022) 122068.
doi: 10.1016/j.fuel.2021.122068
L. Han, X. Liu, J. Chen, et al., Angew. Chem. Int. Ed. 58 (2019) 2321–2325.
doi: 10.1002/anie.201811728
S. Assad, T. Tariq, M. Zaeem Idrees, et al., J. Electroanal. Chem. 931 (2023) 117174.
doi: 10.1016/j.jelechem.2023.117174
M.A. Mushtaq, M. Arif, G. Yasin, et al., Renew. Sust. Energy Rev.176 (2023) 113197.
doi: 10.1016/j.rser.2023.113197
G. Kour, X. Mao, A. Du, J. Mater. Chem. A 10 (2022) 6204–6215.
doi: 10.1039/d1ta08246a
Q. Qin, T. Heil, M. Antonietti, et al., Small Methods. 2 (2018) 1800202.
doi: 10.1002/smtd.201800202
C.Z. He, H. Wang, L.Y. Huai, et al., J. Chem. Phys. 138 (2013) 144703.
doi: 10.1063/1.4798970
Y. Doi, M. Haneda, Catal. Today 303 (2018) 8–12.
doi: 10.1016/j.cattod.2017.07.023
D.L.S. Nieskens, D. Curulla-Ferré, J.W. Niemantsverdriet, ChemPhysChem 7 (2006) 1075–1080.
doi: 10.1002/cphc.200600005
C. He, H. Wang, H. Huai, J. Liu, et al., Chem. J. Chin. Univ. 34 (2013) 946–951.
A.L. Strickler, R.A. Flores, L.A. King, et al., ACS Appl. Mater. Interfaces 11 (2019) 34059–34066.
doi: 10.1021/acsami.9b13697
C.Z. He, H. Wang, P. Zhu, et al., J. Chem. Phys. 135 (2011) 204707.
doi: 10.1063/1.3663621
W. Song, Z. Fu, X. Liu, et al., J. Mater. Chem. A 10 (2022) 13946–13957.
doi: 10.1039/d2ta02642b
W. Song, W. Peng, P. Ma, et al., SSRN Electron. J. 597 (2022) 153678.
C. Wang, B. Yan, J. Zheng, et al., Adv. Powder Mater. 1 (2022) 100018.
doi: 10.1016/j.apmate.2021.11.005
J. Yu, C. He, J. Huo, et al., Int. J. Hydrog. Energy 47 (2022) 7738–7750.
doi: 10.1016/j.ijhydene.2021.12.095
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149–3154.
doi: 10.1016/j.cclet.2021.02.046
D. Ma, W. Ju, Y. Tang, et al., Appl. Surf. Sci. 426 (2017) 244–252.
doi: 10.1016/j.apsusc.2017.07.198
C. He, R. Wang, H. Yang, et al., Appl. Surf. Sci. 507 (2020) 145076.
doi: 10.1016/j.apsusc.2019.145076
J.R. Huo, J. Wang, H.Y. Yang, et al., J. Mol. Model. 27 (2021) 38.
doi: 10.1007/s00894-020-04628-6
C. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci. 509 (2020) 145392.
doi: 10.1016/j.apsusc.2020.145392
G.R. Xu, H. Li, A.S.R. Bati, et al., J. Mater. Chem. A 8 (2020) 15875–15883.
doi: 10.1039/d0ta03237a
M. Gao, Da. Wen, Guo. Cao, et al., Appl. Surf. Sci. 640 (2023) 158286.
doi: 10.1016/j.apsusc.2023.158286
L. Fu, L. Yan, L. Lin, et al., J. Alloys Compd. 875 (2021) 159907.
doi: 10.1016/j.jallcom.2021.159907
F. Rao, G. Zhu, W. Zhang, et al., ACS Catal. 11 (2021) 7735–7749.
doi: 10.1021/acscatal.1c01251
I.A. Erikat, B.A. Hamad, J.M. Khalifeh, Eur. Phys. J. B 87 (2014) 48.
doi: 10.1140/epjb/e2014-40566-x
Z. Zhao, T. Yu, S. Zhang, et al., J. Mater. Chem. A 7 (2019) 405–411.
doi: 10.1039/c8ta09155b
H. Wu, Q.Q. Luo, R.Q. Zhang, et al., Chin. J. Chem. Phys. 31 (2018) 641–648.
doi: 10.1063/1674-0068/31/cjcp1804063
W. Luo, Y. Wang, C. Cheng, Mater. Today Phys. 15 (2020) 100274.
doi: 10.1016/j.mtphys.2020.100274
L. Fu, R. Wang, C.X. Zhao, et al., Chem. Eng. J. 414 (2021) 128857.
doi: 10.1016/j.cej.2021.128857
J. Huo, L. Fu, C. Zhao, et al., Chin. Chem. Lett. 32 (2021) 2269–2273.
doi: 10.1016/j.cclet.2020.12.059
R. Wang, C. He, W. Chen, et al., Chin. Chem. Lett. 32 (2021) 3821–3824.
doi: 10.1016/j.cclet.2021.05.024
J. Li, S. Chen, F. Quan, et al., Chem 6 (2020) 885–901.
doi: 10.3390/polym12040885
V. Rosca, M. Duca, M.T. de Groot, et al., Chem. Rev. 109 (2009) 2209–2244.
doi: 10.1021/cr8003696
S. Licht, B. Cui, B. Wang, et al., Science 345 (2014) 637–640.
doi: 10.1126/science.1254234
X. Lu, J. Zhang, W.K. Chen, et al., Nanoscale Adv. 3 (2021) 1624–1632.
doi: 10.1039/d1na00015b
Z. Huang, M. Rafiq, A.R. Woldu, et al., Coord. Chem. Rev. 478 (2023) 214981.
doi: 10.1016/j.ccr.2022.214981
Y. Ying, K. Fan, X. Luo, et al., Mater. Adv. 1 (2020) 1285–1292.
doi: 10.1039/d0ma00348d
M. Bat-Erdene, G. Xu, M. Batmunkh, et al., J. Mater. Chem. A 8 (2020) 4735–4739.
doi: 10.1039/c9ta13485a
T. Lan, Y. Zhao, J. Deng, et al., Catal. Sci. Technol. 10 (2020) 5792–5810.
doi: 10.1039/d0cy01137a
C. He, J. Wang, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 1051–1057.
doi: 10.1016/j.cclet.2021.09.009
Z. Chen, J. Zhao, C.R. Cabrera, et al., Small Methods 3 (2018) 1800368.
Z. Chen, J. Zhao, L. Yin, et al., J. Mater. Chem. A 7 (2019) 13284–13292.
doi: 10.1039/c9ta01410a
Z.M. Zhang, X. Yao, X.Y. Lang, et al., Appl. Surf. Sci. 536 (2021) 147706.
doi: 10.1016/j.apsusc.2020.147706
C. He, H. Wang, L. Fu, et al., Chin. Chem. Lett. 33(2022) 990–994.
doi: 10.1016/j.cclet.2021.09.049
X. Lv, W. Wei, B. Huang, et al., Nano Lett. 21 (2021) 1871–1878.
doi: 10.1021/acs.nanolett.0c05080
D. Ma, Y. Wang, L. Liu, et al., Phys. Chem. Chem. Phys. 23 (2021) 4018–4029.
doi: 10.1039/d0cp04843g
H. Shen, C. Choi, J. Masa, et al., Chem 7 (2021) 1708–1754.
doi: 10.1016/j.chempr.2021.01.009
X. Zheng, Y. Liu, Y. Yao, Chem. Eng. J. 426 (2021) 130745.
doi: 10.1016/j.cej.2021.130745
Y. Meng, T. Wang, J. Chen, et al., Appl. Surf. Sci. 640 (2023) 158470.
doi: 10.1016/j.apsusc.2023.158470
X. Liu, Y. Geng, R. Hao, et al., Progr. Chem. 33 (2021) 1074–1091.
L. Niu, L. An, X. Wang, et al., J. Energy Chem. 61 (2021) 304–318.
doi: 10.1016/j.jechem.2021.01.018
C. He, R. Sun, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 527–532.
doi: 10.1016/j.cclet.2021.05.072
L. Lin, L. Yan, L. Fu, et al., Fuel 308 (2022) 122068.
doi: 10.1016/j.fuel.2021.122068
L. Han, X. Liu, J. Chen, et al., Angew. Chem. Int. Ed. 58 (2019) 2321–2325.
doi: 10.1002/anie.201811728
S. Assad, T. Tariq, M. Zaeem Idrees, et al., J. Electroanal. Chem. 931 (2023) 117174.
doi: 10.1016/j.jelechem.2023.117174
M.A. Mushtaq, M. Arif, G. Yasin, et al., Renew. Sust. Energy Rev.176 (2023) 113197.
doi: 10.1016/j.rser.2023.113197
G. Kour, X. Mao, A. Du, J. Mater. Chem. A 10 (2022) 6204–6215.
doi: 10.1039/d1ta08246a
Q. Qin, T. Heil, M. Antonietti, et al., Small Methods. 2 (2018) 1800202.
doi: 10.1002/smtd.201800202
C.Z. He, H. Wang, L.Y. Huai, et al., J. Chem. Phys. 138 (2013) 144703.
doi: 10.1063/1.4798970
Y. Doi, M. Haneda, Catal. Today 303 (2018) 8–12.
doi: 10.1016/j.cattod.2017.07.023
D.L.S. Nieskens, D. Curulla-Ferré, J.W. Niemantsverdriet, ChemPhysChem 7 (2006) 1075–1080.
doi: 10.1002/cphc.200600005
C. He, H. Wang, H. Huai, J. Liu, et al., Chem. J. Chin. Univ. 34 (2013) 946–951.
A.L. Strickler, R.A. Flores, L.A. King, et al., ACS Appl. Mater. Interfaces 11 (2019) 34059–34066.
doi: 10.1021/acsami.9b13697
C.Z. He, H. Wang, P. Zhu, et al., J. Chem. Phys. 135 (2011) 204707.
doi: 10.1063/1.3663621
W. Song, Z. Fu, X. Liu, et al., J. Mater. Chem. A 10 (2022) 13946–13957.
doi: 10.1039/d2ta02642b
W. Song, W. Peng, P. Ma, et al., SSRN Electron. J. 597 (2022) 153678.
C. Wang, B. Yan, J. Zheng, et al., Adv. Powder Mater. 1 (2022) 100018.
doi: 10.1016/j.apmate.2021.11.005
J. Yu, C. He, J. Huo, et al., Int. J. Hydrog. Energy 47 (2022) 7738–7750.
doi: 10.1016/j.ijhydene.2021.12.095
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149–3154.
doi: 10.1016/j.cclet.2021.02.046
D. Ma, W. Ju, Y. Tang, et al., Appl. Surf. Sci. 426 (2017) 244–252.
doi: 10.1016/j.apsusc.2017.07.198
C. He, R. Wang, H. Yang, et al., Appl. Surf. Sci. 507 (2020) 145076.
doi: 10.1016/j.apsusc.2019.145076
J.R. Huo, J. Wang, H.Y. Yang, et al., J. Mol. Model. 27 (2021) 38.
doi: 10.1007/s00894-020-04628-6
C. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci. 509 (2020) 145392.
doi: 10.1016/j.apsusc.2020.145392
G.R. Xu, H. Li, A.S.R. Bati, et al., J. Mater. Chem. A 8 (2020) 15875–15883.
doi: 10.1039/d0ta03237a
M. Gao, Da. Wen, Guo. Cao, et al., Appl. Surf. Sci. 640 (2023) 158286.
doi: 10.1016/j.apsusc.2023.158286
L. Fu, L. Yan, L. Lin, et al., J. Alloys Compd. 875 (2021) 159907.
doi: 10.1016/j.jallcom.2021.159907
F. Rao, G. Zhu, W. Zhang, et al., ACS Catal. 11 (2021) 7735–7749.
doi: 10.1021/acscatal.1c01251
I.A. Erikat, B.A. Hamad, J.M. Khalifeh, Eur. Phys. J. B 87 (2014) 48.
doi: 10.1140/epjb/e2014-40566-x
Z. Zhao, T. Yu, S. Zhang, et al., J. Mater. Chem. A 7 (2019) 405–411.
doi: 10.1039/c8ta09155b
H. Wu, Q.Q. Luo, R.Q. Zhang, et al., Chin. J. Chem. Phys. 31 (2018) 641–648.
doi: 10.1063/1674-0068/31/cjcp1804063
W. Luo, Y. Wang, C. Cheng, Mater. Today Phys. 15 (2020) 100274.
doi: 10.1016/j.mtphys.2020.100274

Yuxiang Zhang , Jia Zhao , Sen Lin . Nitrogen doping retrofits the coordination environment of copper single-atom catalysts for deep CO2 reduction. Chinese Journal of Structural Chemistry, 2024, 43(11): 100415-100415. doi: 10.1016/j.cjsc.2024.100415
Chaozheng He , Jia Wang , Ling Fu , Wei Wei . Nitric oxide assists nitrogen reduction reaction on 2D MBene: A theoretical study. Chinese Chemical Letters, 2024, 35(5): 109037-. doi: 10.1016/j.cclet.2023.109037
Xu Huang , Kai-Yin Wu , Chao Su , Lei Yang , Bei-Bei Xiao . Metal-organic framework Cu-BTC for overall water splitting: A density functional theory study. Chinese Chemical Letters, 2025, 36(4): 109720-. doi: 10.1016/j.cclet.2024.109720
Wei Chen , Pieter Cnudde . A minireview to ketene chemistry in zeolite catalysis. Chinese Journal of Structural Chemistry, 2024, 43(11): 100412-100412. doi: 10.1016/j.cjsc.2024.100412
Jiajun Wang , Guolin Yi , Shengling Guo , Jianing Wang , Shujuan Li , Ke Xu , Weiyi Wang , Shulai Lei . Computational design of bimetallic TM2@g-C9N4 electrocatalysts for enhanced CO reduction toward C2 products. Chinese Chemical Letters, 2024, 35(7): 109050-. doi: 10.1016/j.cclet.2023.109050
Weiping Xiao , Yuhang Chen , Qin Zhao , Danil Bukhvalov , Caiqin Wang , Xiaofei Yang . Constructing the synergistic active sites of nickel bicarbonate supported Pt hierarchical nanostructure for efficient hydrogen evolution reaction. Chinese Chemical Letters, 2024, 35(12): 110176-. doi: 10.1016/j.cclet.2024.110176
Mianfeng Li , Haozhi Wang , Zijun Yang , Zexiang Yin , Yuan Liu , Yingmei Bian , Yang Wang , Xuerong Zheng , Yida Deng . Synergistic enhancement of alkaline hydrogen evolution reaction by role of Ni-Fe LDH introducing frustrated Lewis pairs via vacancy-engineered. Chinese Chemical Letters, 2025, 36(3): 110199-. doi: 10.1016/j.cclet.2024.110199
Fanjun Kong , Yixin Ge , Shi Tao , Zhengqiu Yuan , Chen Lu , Zhida Han , Lianghao Yu , Bin Qian . Engineering and understanding SnS0.5Se0.5@N/S/Se triple-doped carbon nanofibers for enhanced sodium-ion batteries. Chinese Chemical Letters, 2024, 35(4): 108552-. doi: 10.1016/j.cclet.2023.108552
Yu-Hang Li , Shuai Gao , Lu Zhang , Hanchun Chen , Chong-Chen Wang , Haodong Ji . Insights on selective Pb adsorption via O 2p orbit in UiO-66 containing rich-zirconium vacancies. Chinese Chemical Letters, 2024, 35(8): 109894-. doi: 10.1016/j.cclet.2024.109894
Yuwei Liu , Yihui Zhu , Weijian Duan , Yizhuo Yang , Haorui Tuo , Chunhua Feng . Electrocatalytic nitrate reduction on Fe, Fe3O4, and Fe@Fe3O4 cathodes: Elucidating structure-sensitive mechanisms of direct electron versus hydrogen atom transfer. Chinese Chemical Letters, 2025, 36(6): 110347-. doi: 10.1016/j.cclet.2024.110347
Lingling Su , Qunyan Wu , Congzhi Wang , Jianhui Lan , Weiqun Shi . Theoretical design of polyazole based ligands for the separation of Am(Ⅲ)/Eu(Ⅲ). Chinese Chemical Letters, 2024, 35(8): 109402-. doi: 10.1016/j.cclet.2023.109402
Xin-Tong Zhao , Jin-Zhi Guo , Wen-Liang Li , Jing-Ping Zhang , Xing-Long Wu . Two-dimensional conjugated coordination polymer monolayer as anode material for lithium-ion batteries: A DFT study. Chinese Chemical Letters, 2024, 35(6): 108715-. doi: 10.1016/j.cclet.2023.108715
Ze Zhang , Lei Yang , Jin-Ru Liu , Hao Hu , Jian-Li Mi , Chao Su , Bei-Bei Xiao , Zhi-Min Ao . Improved oxygen electrocatalysis at FeN4 and CoN4 sites via construction of axial coordination. Chinese Chemical Letters, 2025, 36(2): 110013-. doi: 10.1016/j.cclet.2024.110013
Teng Wang , Jiachun Cao , Juan Li , Didi Li , Zhimin Ao . A novel photocatalytic mechanism of volatile organic compounds degradation on BaTiO3 under visible light: Photo-electrons transfer from photocatalyst to pollutant. Chinese Chemical Letters, 2025, 36(3): 110078-. doi: 10.1016/j.cclet.2024.110078
Yubang Liu , Jiaxin Lin , Huayu Liang , Yinwu Li , Zhuofeng Ke . Computational insights into three-centre four-electron bridging hydride bond in boryl type PBP-M dihydride complexes✰ ✩. Chinese Chemical Letters, 2025, 36(5): 110291-. doi: 10.1016/j.cclet.2024.110291
Maitri Bhattacharjee , Rekha Boruah Smriti , R. N. Dutta Purkayastha , Waldemar Maniukiewicz , Shubhamoy Chowdhury , Debasish Maiti , Tamanna Akhtar . Synthesis, structural characterization, bio-activity, and density functional theory calculation on Cu(Ⅱ) complexes with hydrazone-based Schiff base ligands. Chinese Journal of Inorganic Chemistry, 2024, 40(7): 1409-1422. doi: 10.11862/CJIC.20240007
Ling Tang , Yan Wan , Yangming Lin . Lowering the kinetic barrier via enhancing electrophilicity of surface oxygen to boost acidic oxygen evolution reaction. Chinese Journal of Structural Chemistry, 2024, 43(11): 100345-100345. doi: 10.1016/j.cjsc.2024.100345
Ting Xie , Xun He , Lang He , Kai Dong , Yongchao Yao , Zhengwei Cai , Xuwei Liu , Xiaoya Fan , Tengyue Li , Dongdong Zheng , Shengjun Sun , Luming Li , Wei Chu , Asmaa Farouk , Mohamed S. Hamdy , Chenggang Xu , Qingquan Kong , Xuping Sun . CoSe2 nanowire array enabled highly efficient electrocatalytic reduction of nitrate for ammonia synthesis. Chinese Chemical Letters, 2024, 35(11): 110005-. doi: 10.1016/j.cclet.2024.110005
Zhengkun QIN , Zicong PAN , Hui TIAN , Wanyi ZHANG , Mingxing SONG . A series of iridium(Ⅲ) complexes with fluorophenyl isoquinoline ligand and low-efficiency roll-off properties: A density functional theory study. Chinese Journal of Inorganic Chemistry, 2025, 41(6): 1235-1244. doi: 10.11862/CJIC.20240429
Hang Wang , Qi Wang , Chuan-De Wu . Continuous synthesis of ammonia. Chinese Journal of Structural Chemistry, 2025, 44(3): 100437-100437. doi: 10.1016/j.cjsc.2024.100437
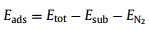
 DownLoad:
DownLoad:
 DownLoad:
DownLoad: