Citation:
Yan-Bo Li, Yi Li, Liang Yin. Copper(Ⅰ)-catalyzed diastereodivergent construction of vicinal P-chiral and C-chiral centers facilitated by dual "soft-soft" interaction[J]. Chinese Chemical Letters,
;2024, 35(7): 109294.
doi:
10.1016/j.cclet.2023.109294
Copper(Ⅰ)-catalyzed diastereodivergent construction of vicinal P-chiral and C-chiral centers facilitated by dual "soft-soft" interaction
CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Centre for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200032, China
A copper(Ⅰ)-catalyzed diastereodivergent addition of phosphinothioates (HP(S)ROR') to α, β-unsaturated thioamides is disclosed, which constructs vicinal P-chiral and C-chiral centers in generally high diastereo- and enantioselectivities. In this reaction, the kinetic resolution of HP(S)ROR' occurs, which affords (R)-HP(S)PhOMe in high enantioselectivity in the addition with (R, R)-Ph-BPE as the ligand. It is found through control experiment that dual "soft-soft" interaction, indicated by both 1H and 31P NMR experiments, is indispensable in the present reaction. The first "soft-soft" interaction between copper(Ⅰ) catalyst and HP(S)ROR' enables facile deprotonation to generate nucleophilic [Cu]-SPROR' species. The second one between the [Cu]-SPROR' species and α, β-unsaturated thioamides facilitated the nucleophilic addition. Finally, both Michael adducts and (R)-HP(S)PhOMe are easily converted to synthetically useful compounds.
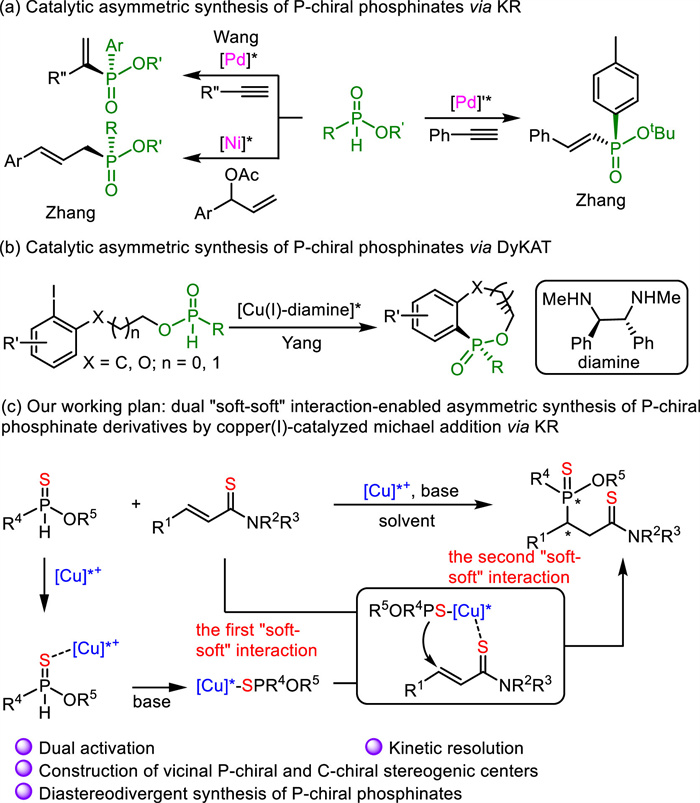
Recently, there has been a considerable interest in the asymmetric synthesis of P-chiral compounds as P-chiral stereogenic centers appear as key structural features not only in bioactive molecules [1,2] but also in chiral ligands [3–6]. Besides the classical methods based on transformations from chiral pool or auxiliary-assisted diastereoselective synthesis [7,8], new methods focusing on asymmetric catalysis contribute to the efficient asymmetric synthesis of P-chiral compounds [9–12]. Among P-chiral compounds, phosphinates are a class of important compounds in both medicinal and synthetic chemistry. For example, several chiral phosphinates exhibit strong anti-proliferative activity against a large panel of NCI cancer cell lines [13]. Furthermore, it is well known that chiral phosphinates serve as starting materials for the preparation of P-chiral tertiary phosphine oxides [14]. However, there are only limited methods available for the catalytic asymmetric preparation of P-chiral phosphinates.
Three strategies have been employed for the catalytic asymmetric synthesis of P-chiral phosphinates. The first one is the catalytic desymmetrization. Both transition metal catalysis and organocatalysis enabled the facile synthesis of P-chiral phosphinates via desymmetrization [15–18]. The second one is the catalytic kinetic resolution (KR) (Scheme 1a). In 2020, both Wang and Zhang studied the construction of P-chiral phosphinates by means of palladium-catalyzed asymmetric hydrophosphorylation of alkynes via KR [19,20]. Zhang's method was particularly efficient for secondary phosphine oxides but not very suitable for H-phosphinate. Moreover, Zhang and co-workers developed a Ni-catalyzed enantioselective allylic alkylation of H-phosphinates via KR based on their previous work with secondary phosphine oxides [21–23]. The third one is catalytic dynamic kinetic asymmetric transformation (DyKAT) (Scheme 1b). In 2022, Yang and co-workers disclosed a copper(Ⅰ)-catalyzed dynamic kinetic intramolecular C-P cross-coupling reaction, which delivered P-chiral cyclic phosphinates in high enantioselectivity [24]. Despite the above achievements via three strategies, new methods towards the catalytic asymmetric synthesis of more diversified P-chiral phosphinates remain elusive. Conjugate addition has been identifed as a powerful tool to access P-chiral phosphine derivatives [25–34]. However, its application in the synthesis of P-chiral phosphinates via KR has never been disclosed largely due to the weak nucleophicility of H-phosphinates [21].
Scheme 1
Scheme 1.
Catalytic asymmetric synthesis of P-chiral phosphinates via KR or DyKAT and our working hypothesis.
The catalytic asymmetric construction of vicinal tertiary and quaternary stereogenic centers is a challenging issue as there are limited strateties available in literature. The possbile reasons inlcude difficult diastereo- and enantio-discrimination caused by the rotational freedom of acyclic molecules and steric repulsion caused by the two multi-substituted stereogenic centers [35–38]. It would be more challenging to achieve the construction of vicinal P-chiral center and C-chiral center through asymmetric catalysis, which is reflected by the very limited methodologies [25–30,39–41]. In such reported reactions, the pronucleophiles were limited to racemic secondary phosphines, which led to a DyKAT process. Moreover, the diastereodivergent synthesis [42,43] of each diastereoisomer has never been achieved. Herein, by means of the dual "soft-soft" interaction [44], a copper(Ⅰ)-catalyzed diastereodivergent construction of vicinal P-chiral center and C-chiral center was accomplished, providing new chiral phosphinate derivatives in generally high to excellent diastereo- and enantioselectivities (Scheme 1c). Interestingly, a kinetic resolution process accompanies the catalytic asymmetric Michael addition. Moreover, the dual "soft-soft" interaction between copper(Ⅰ) and sulfur-containing nucleophiles (phosphinothioates), as well as electrophiles (α, β-unsaturated thioamides [45–47, [48]), was carefully studied through NMR analysis.
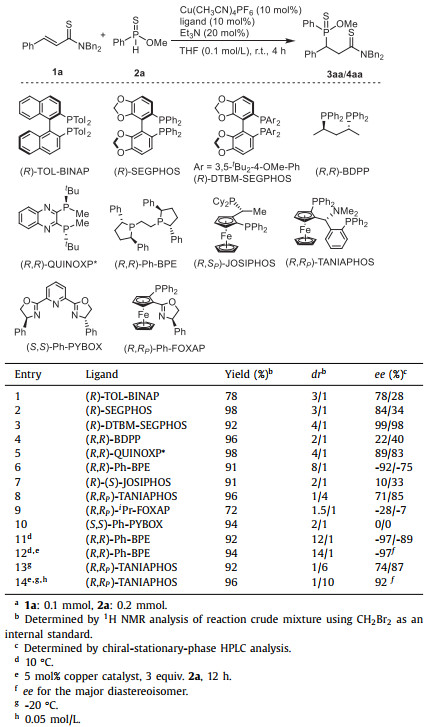
The reaction of α, β-unsaturated thioamide 1a and O-methyl phenylphosphinothioate (2a) was studied for the optimization of reaction conditions (Table 1). In the presence of 10 mol% Cu(CH3CN)4PF6, 10 mol% (R)-TOL-BINAP, and 20 mol% Et3N, the conjugate addition with 1.0 equiv. 1a and 2.0 equiv. 2a occurred smoothly to deliver the product 3aa in moderate yield with unsatisfied diastereo- and enantioselectivities in 4 h (entry 1). Then, a series of bisphosphine ligands were further evaluated (entry 2-7). The highest diastereoselectivity was observed in the reaction with (R, R)-Ph-BPE (entry 6). Fortunately, the enantioselectivity for the major diastereoisomer 3aa was high. Interestingly, in the reaction with (R, R)-TANIAPHOS, the diastereoselectivity was reversed (entry 8). Instead of 3aa, its diastereoisomer (4aa) was observed in 4/1 dr with 85%/71% ee. Except for the bisphosphine ligands, (R, R)-iPr-FOXAP and (S, S)-Ph-PYBOX were also tested. However, no superior results were obtained (entries 9 and 10). By setting up the reaction with (R, R)-Ph-BPE at 10 ℃ and increasing the amount of 2a to 3.0 equiv., 3aa was afforded in enhanced both diastereo- and enantioselectivities in 12 h (entry 12). As for the reaction with (R, R)-TANIAPHOS, 4aa was generated in 10/1 dr with 92% ee at ‒20 ℃ in the presence of 3.0 equiv. 2a with a concentration of 0.05 mol/L for 1a (entry 14).
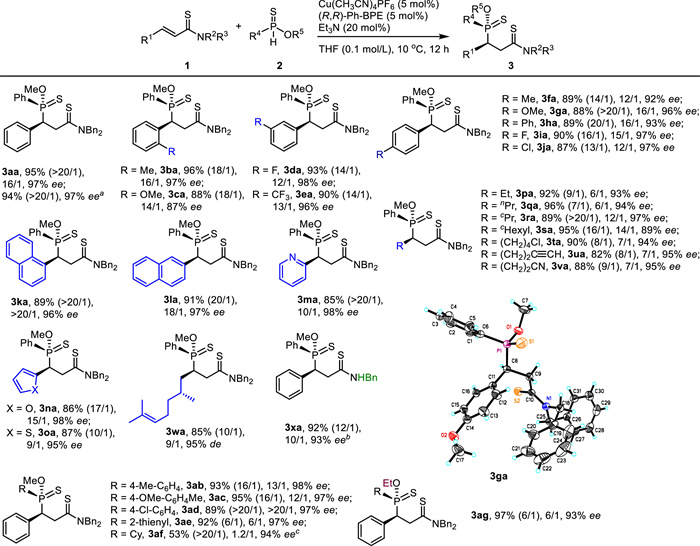
By using the reaction conditions in entry 12 in Table 1, the substrate scopes for synthesizing 3 were evaluated as given in Scheme 2. It should be noticed that the dr values were generally enhanced in the isolation process. As for R1, various mono-substituted phenyls were applied. The excellent reaction results (3ba-3ja, 87%-96%, 13/1 ~ > 20/1 dr, 87%-98% ee) were not affected by the substitution pattern on the phenyl ring, including the ortho-, meta-, and para-substitution. Both electron-donating groups and electron-withdrawing groups were also well tolerated on the phenyl group. Moreover, both 1-naphthyl and 2-naphthyl were nicely accepted at the R1 position with satisfactory results obtained (3ka-3la, 89%-91%, 20/1 ~ > 20/1 dr, 96%-97% ee). Some heteroaryl groups, such as 2-pyridinyl, 2-furanyl, and 2-thienyl were also appropriate for R1 (3ma-3oa, 85%-87%, 10/1 ~ > 20/1 dr, 95%-98% ee).
Scheme 2
Scheme 2.
Substrate scope I of the catalytic asymmetric reaction. 1: 0.2 mmol, 2: 0.6 mmol. Isolated yields. ee of the major diastereoisomer was determined by chiral-stationary-phase HPLC analysis. dr was determined by 1H NMR analysis of crude mixture. dr in parenthesis was determined by 1H NMR analysis of an isolated sample. a Gram-scale reaction. b 10 mol% copper(Ⅰ) catalyst used. c 10 mol% Cu(CH3CN)4PF6 and 10 mol% (R)-DTBM-SEGPHOS used at room temperature.
Then several alkyl groups for R1 were tested. It was found that simple linear alkyls, simple cyclic alkyls, and functionalized alkyls were well applicable (3pa-3va, 82%-96%, 7/1 ~ > 20/1 dr, 89%-97% ee). It was notable that alkyl chloride was not touched by the pronucleophile 2a. Furthermore, the terminal alkyne group did not disturb the addition although alkynyl copper(Ⅰ) species was often formed, which might undergo a conjugate addition to unsaturated amide 1a [42]. The reaction results were not disturbed by the presence of a nearby chiral center (3wa, 85%, 10/1 dr, 95% de). Interestingly, an α, β-unsaturated N-H thioamide served as a wonderful electrophile (3xa, 92%, 12/1 dr, 93% ee).
The phenylphosphinothioate 2a was then varied. 4-Methyl-phenyl, 4-OMe-phenyl, 4-Cl-phenyl, and 2-thienyl were introduced into R4. The corresponding products were isolated in good results (3ab-3ae, 89%-95%, 6/1 ~ > 20/1 dr, 97%-98% ee). However, the phosphinothioate with cyclohexyl at R4 position worked in the present conditions with low diastereoselectivity (3af, 1.2/1 dr). Fortunately, 3af was isolated in moderate yield with excellent dr (53%, > 20/1 dr, 94% ee). O-Ethyl phenylphosphinothioate (2g) was also a good substrate (3ag, 97%, 6/1 dr, 94% ee). The absolute configuration of 3ga was determined by X-ray analysis and the stereochemistry of other products (3) was deduced by the structural analogy.
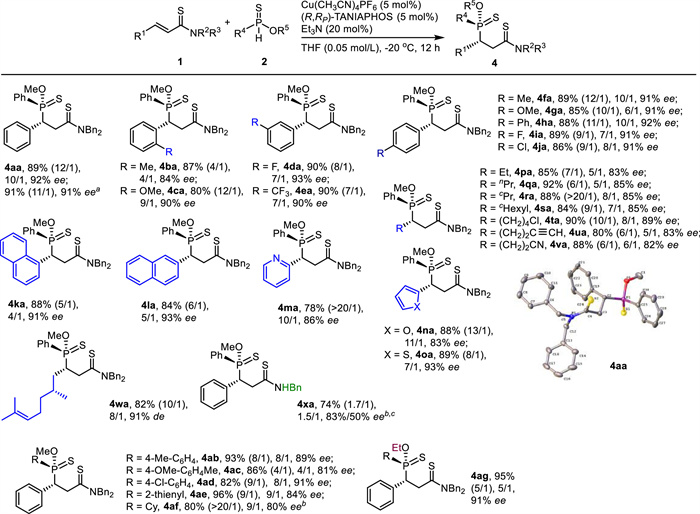
By using the reaction conditions in entry 14 in Table 1, both unsaturated amides 1 and phosphinothioates 2 evaluated in Scheme 2 were examined again to assemble 4 (Scheme 3). As for α, β-unsaturated amides 1, both aryls and heteroaryls were suitable substituents for R1 as the corresponding products were isolated in high yields with moderate to high diastereo- and high enantioselectivity (4aa-4oa, 78%-90%, 4/1 ~ > 20/1 dr, 83%-93% ee). Various alkyls served as proper substituents at R1 too (4pa-4va, 80%-92%, 6/1 ~ > 20/1 dr, 82%-89% ee). The α, β-unsaturated thioamide bearing a chiral center (1w) worked also as a suitable electrophile (4wa, 82%, 10/1 dr, 91% de). However, the reaction of α, β-unsaturated N-H thioamide 2x occurred in moderate yield with 1.7/1 dr and 83%/50% ee (4xa, 74%). The bad dr was attributed to the decreased steric hindrance of N-H thioamide. As for phosphinothioates 2, both aryls and heteroaryl were well accepted at R4 (4ab-4ae, 82%-96%, 4/1-9/1 dr, 81%-91% ee). Remarkably, O-methyl cyclohexylphosphinothioate (2f) underwent the Michael addition to give 4af in satisfactory results (4af, 80%, > 20/1 dr, 80% ee). Although the enantioselectivity was lower than the one in Scheme 2, the diastereoselectivity in the crude mixture was significantly increased (9/1 vs. 1.2/1). The reaction with O-ethyl phenylphosphinothioate (2g) also worked nicely to deliver the desired product (4ag, 95%, 5/1 dr, 91% ee). Generally speaking, both the diastereo- and the enantioselectivities obtained in Scheme 3 were lower than the ones acquired in Scheme 2. The absolute configuration of 4aa was determined unambiguously with the assistance of X-ray crystallography. The stereochemistry in other products (4) was tentatively assigned.
Scheme 3
Scheme 3.
Substrate scope II of the catalytic asymmetric reaction. 1: 0.2 mmol, 2: 0.6 mmol. Isolated yields. ee of the major diastereoisomer was determined by chiral-stationary-phase HPLC analysis. dr was determined by 1H NMR analysis of crude mixture (in parenthesis of an isolated sample). a Gram-scale reaction. b 10 mol% catalyst loading. c 24 h.
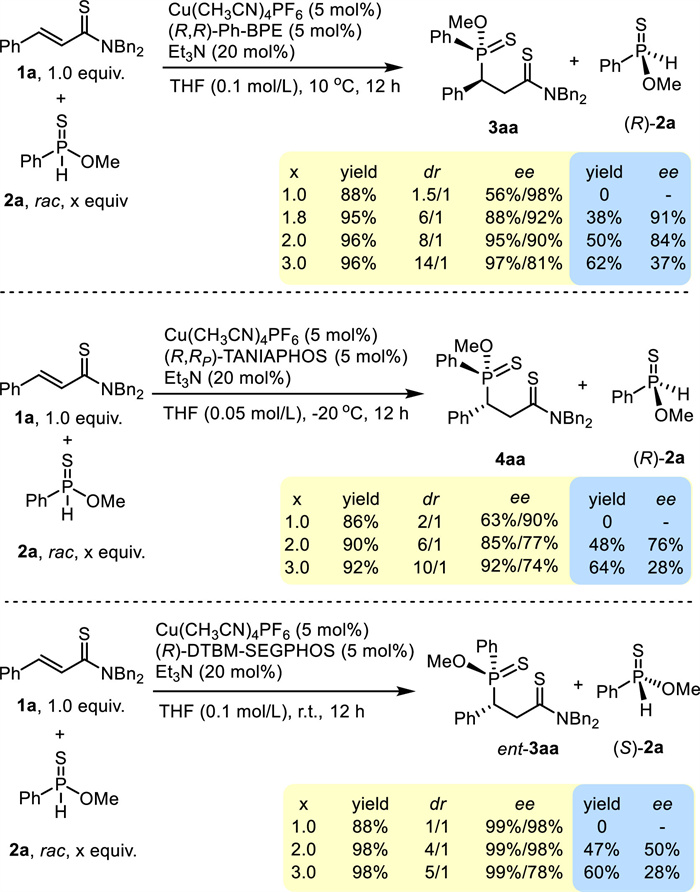
After the studies on the substrate scope, the kinetic resolution of 2a was investigated. As shown in Scheme 4, with (R, R)-Ph-BPE as the ligand, the reaction with 1.0 equiv. 1a and 1.0 equiv. rac-2a afforded 3aa in 88% yield with 1.5/1 dr and 56%/98% ee. By increasing to 1.8 equiv. rac-2a, the reaction furnished 3aa in 95% yield with 6/1 dr and 88%/92% ee. Meanwhile, (R)-2a was obtained in 38% yield with 91% ee. With 2.0 equiv. rac-2a, both the dr and the ee of the major diastereoisomer increased (8/1 dr, 95%/90% ee). (R)-2a was generated in 50% yield with 84% ee. When 3.0 equiv. rac-2a was employed, 3aa was generated in 96% yield with 14/1 dr and 97%/81% ee and (R)-2a was recovered in 62% yield with 37% ee. Then, the kinetic resolution of 2a with (R, R)-TANIAPHOS was probed. The reaction with 1.0 equiv. 1a and 1.0 equiv. rac-2a afforded 4aa in 86% yield with 2/1 dr and 63%/90% ee. In the reaction with 1.0 equiv. 1a and 2.0 equiv. rac-2a, (R)-2a was afforded in 48% yield with 76% ee. The reaction with 3.0 equiv. rac-2a gave 4aa in 92% yield with 10/1 dr and 92%/74% ee and 64% (R)-2a was obtained in 28% ee. Evidently, the resolution of 2a with (R, R)-Ph-BPE is more efficient than the one with (R, R)-TANIAPHOS. It was noted that the reaction with (R)-DTBM-SEGPHOS gave the corresponding adduct in excellent enantioselectivity for both diastereoisomers (Table 1, entry 3). Therefore, the dynamic resolution with (R)-DTBM-SEGPHOS was also tested. In the reaction with 1.0 equiv. 1a and 1.0 equiv. rac-2a, two diastereoisomers with 1/1 ratio were obtained in a total yield of 88%. By using 2.0 equiv. 2a, the reaction provided both ent-3aa in 98% yield with 4/1 dr and 99%/98% ee and (S)-2a in 47% yield with 50% ee. It is deduced that ent-3aa and the major enantiomer in its diastereoisomers had reversed P-chiral centers, which would rationalize the above experimental facts.
Scheme 4
Scheme 4.
Control experiments on kinetic resolution of 2a.
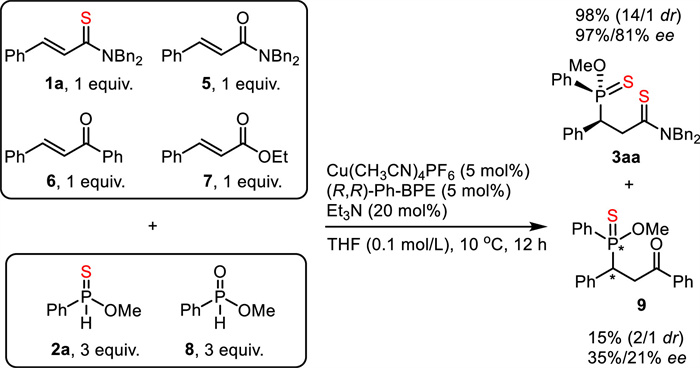
The power of dual "soft-soft" interaction was demonstrated by a competitive experiment (Scheme 5). A test tube was charged with 5 mol% Cu(CH3CN)4PF6, 5 mol% (R, R)-Ph-BPE, 20 mol% Et3N, and THF ahead. Then four electrophiles (1.0 equiv. for each), including unsaturated thioamide 1a, unsaturated amide 5, unsaturated ketone 6, and unsaturated ester 7, and two nucleophiles (3.0 equiv. for each), including methyl phenylphosphinate 8 and O-methyl phenylphosphinothioate 2a were added to the test tube. After stirring the reaction mixture at 10 ℃ for 12 h, 3aa was obtained in 98% yield with 14/1 dr and 97% ee together with 9 in 15% yield. It should be noted that 9 was generated in low yield, low dr, and low enantioselectivity. Obviously, under the "soft-soft" interaction, α, β-unsaturated thioamide exhibited even higher electrophilicity than unsaturated ketone. Furthermore, O-methyl phenylphosphinothioate showed much higher reactivity than methyl phenylphosphinate. Evidently, this experimental fact demonstrated the unique advantage of the dual "soft-soft" interaction in transition-metal catalysis.
Scheme 5
Scheme 5.
Competitive experiment (1H NMR yields based on 1a and 6 are given).
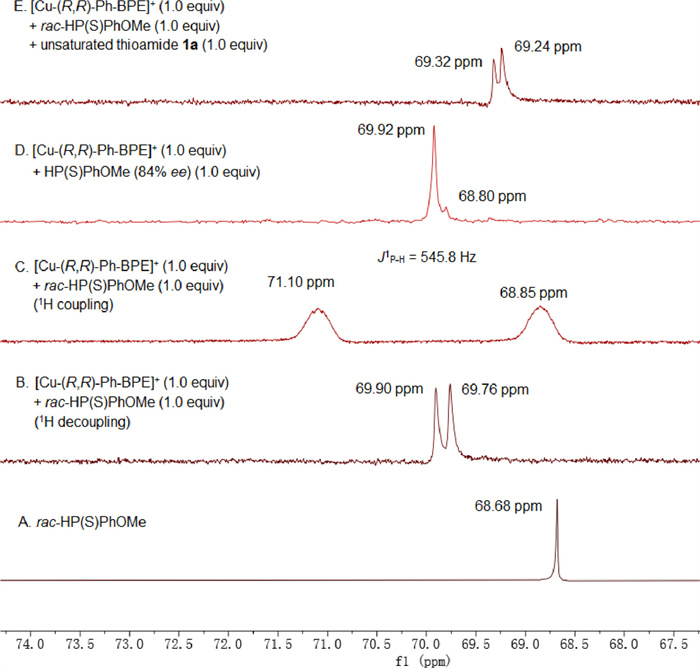
Then, some NMR studies were performed to get insights into the dual "soft-soft" interaction as shown in Fig. 1 (for the full spectra among the range of -5~100 ppm, see Supporting information). The 31P signal of rac-HP(S)PhOMe was recorded as δ 68.68 ppm (Fig. 1A). The 1/1 mixture of [Cu‒(R, R)-Ph-BPE]+ and rac-HP(S)PhOMe gave two 31P signals at δ 69.90 and δ 69.76 ppm with 1/1 dr ratio in the range of 66-75 ppm (Fig. 1B), which indicated the formation of [Cu‒(R, R)-Ph-BPE]+‒rac-HP(S)PhOMe species and thus confirmed the "soft-soft" interaction between copper(Ⅰ) and HP(S)PhOMe. Evidently, the 1/1 dr ratio is in accordance with the fact that rac-HP(S)PhOMe was employed. In the spectrum without decoupling by 1H (Fig. 1C), two peaks with a J = 545.8 Hz were observed. The huge H-P coupling constant implied that P(V) was present in the complex rather than HSP(Ⅲ)PhOMe. By using a sample of (R)-HP(S)PhOMe with 84% ee (Fig. 1D), the appearances of a big peak at δ 69.92 ppm and a small peak at δ 68.80 ppm further confirmed the formation of the [Cu‒(R, R)-Ph-BPE]+‒HP(S)PhOMe complex. Finally, the 1/1/1 mixture of [Cu‒(R, R)-Ph-BPE]+, rac-HP(S)PhOMe, unsaturated thioamide 1a gave two 31P signals at δ 69.32 and δ 69.24 ppm, which might suggest the formation of the [Cu‒(R, R)-BPE]+‒rac-HP(S)PhOMe‒thioamide 1a complex (Fig. 1E).
In order to further explore the "soft-soft" interaction between copper(Ⅰ)-bisphosphine complex and α, β-unsaturated thioamide 1a, the 1H NMR spectrum of the 1/1 mixture of [Cu‒(R)-BINAP]+ and 1a was studied (for the details, see Supporting information). Although the chemical shifts of the two benzylic methylenes in the mixture were almost the same as the counterparts in 1a, the four methylene hydrogens in the mixture gave doublet peaks, which were different from the two single peaks in 1a. This information indicated that there was a chiral environment around the two hydrogens in the methylenes and thus the [Cu‒(R)-BINAP]+-1a complex was formed, which provided the required chiral environment. Evidently, there is a "soft-soft" interaction between the [Cu‒(R)-BINAP]+ complex and α, β-unsaturated thioamide 1a. In the study of the 1/1/1 mixture of [Cu‒(R)-BINAP]+, 1a, and 2a, a similar instructive phenomenon was observed (for the details, see Supporting information), which also indicated the "soft-soft" interaction between the [Cu‒(R)-BINAP]+-2a complex and α, β-unsaturated thioamide 1a. Evidently, these NMR experiments offered some proof for the double "soft-soft" interaction.
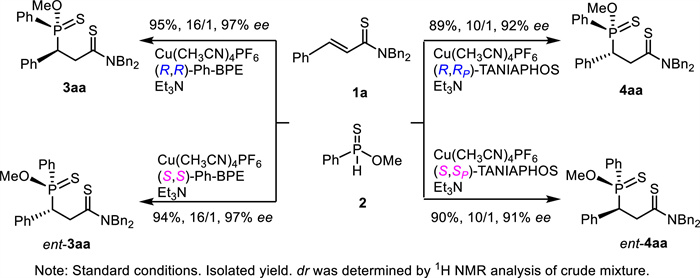
The stereodivergent synthesis of all four stereoisomers was easily achieved by using unsaturated thioamide 1a and O-methyl phenylphosphinothioate (2a) as model substrates (Scheme 6). With (S, S)-Ph-BPE instead of (R, R)-Ph-BPE, the reaction proceeded in the same reaction efficiency, affording ent-3aa in 94% yield with 16/1 dr and 97% ee. By replacing (R, R)-TANIAPHOS with (S, S)-TANIAPHOS, ent-4aa was prepared in 90% yield with 10/1 dr and 91% ee. Evidently, both (R, R)-Ph-BPE and (R, R)-TANIAPHOS led to P-stereogenic center with (S)-configuration while both (S, S)-Ph-BPE and (S, S)-TANIAPHOS led to P-stereogenic center with (R)-configuration.
Scheme 6
Scheme 6.
Stereodivergent synthesis to all four stereoisomers with vicinal P-chiral and C-chiral centers.
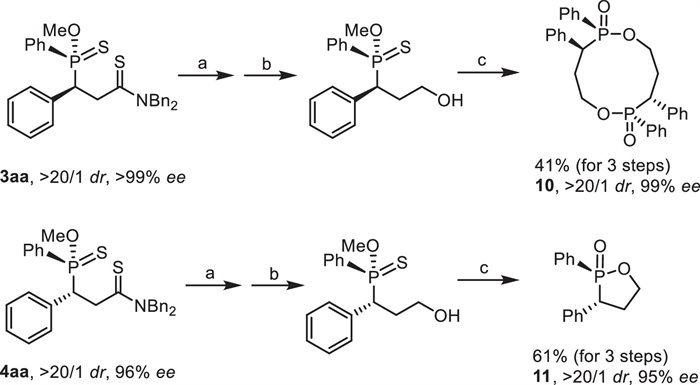
Subsequently, transformations of the products were proved to be straightforward by using 3aa and 4aa as model substrates (Scheme 7). By reduction with Schwartz reagent [49] at –78 ℃ and further reduction with NaBH4, thioamide 3aa was converted to a chiral alcohol, which was then treated with mCPBA. Interestingly, an intermolecular esterification occurred to give cyclic diphosphinate 10 in 41% yield for three steps with uncompromising both diastereo- and enantioselectivities. It should be noted that without the oxidative conversion of phosphinothioate to phosphinate, the intermolecular esterification did not proceed at all. Furthermore, the P-chiral centers were reversed in the esterification. On the contrary, the three-step transformation of 4aa afforded cyclic monophosphinate 11 rather than diphosphinate in 61% yield with unchanged both dr and ee by an intramolecular esterification. This special phenomenon highlighted the potential different reaction behavior of two diastereoisomers under the same reaction condition. At this stage, the origin for this interesting phenomenon is unclear.
Scheme 7
Scheme 7.
Transformations of 3aa and 4aa. Conditions: (a) Cp2ZrHCl, toluene, -78 ℃ to r.t. (b) NaBH4, MeOH, r.t. (c) mCPBA, DCM, 0 ℃ to r.t.
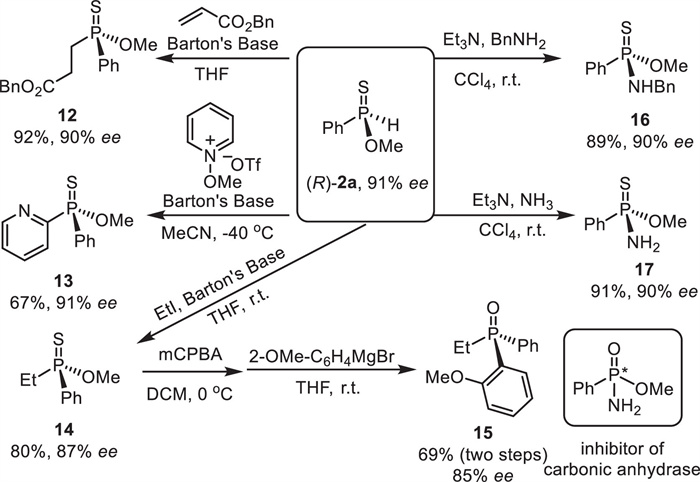
The synthetic utility of chiral O-methyl phenylphosphinothioate ((R)-2a) was demonstrated by several transformations (Scheme 8). The conjugate addition of (R)-2a to benzyl acrylate was successfully carried out, furnishing thiophosphinate 12 in 92% yield with uncompromising ee. Moreover, the heteroarylation reaction of (R)-2a through nucleophilic aromatic substitution proceeded smoothly at ‒40 ℃ to deliver thiophosphinate 13 in 67% yield with unchanged ee [50]. The alkylation with EtI worked well to produce thiophosphinate 14 in 80% yield with slightly decreased ee. 14 was easily converted to tertiary phosphine oxide 15 in 69% yield for two steps with slightly eroded ee [14]. Notably, 15 is easily reduced to P-chiral tertiary phosphine, which may serve as chiral ligands in transition-metal catalysis. Finally, an amination with benzylamine by means of the Atherton-Todd reaction [51] led to phosphonamidothioate 16 in 89% yield with remained ee. Ammonia also served as a wonderful substrate in the amination to give phosphonamidothioate 17 in 91% yield with retained ee. It should be noted that the P-chiral centers were reversed in the Atherton-Todd reaction. Moreover, it is interesting to see that the oxide derivative of 17 was identified as an inhibitor of carbonic anhydrase [52,53].
In conclusion, with the assistance of the dual "soft-soft" interaction between copper(Ⅰ) and the sulfur atoms in substrates, a catalytic asymmetric diastereodivergent addition of phosphinothioates to α, β-unsaturated thioamides was successfully carried out. The diastereodivergent additions were achieved by using (R, R)-Ph-BPE and (R, R)-TANIAPHOS as the ligand, respectively. The Michael addition enjoyed broad substrate scopes on both α, β-unsaturated thioamides and phosphinothioates. In the meanwhile of asymmetric addition, the kinetic resolution of phosphinothioates occurred. With (R, R)-BPE as the ligand, the resolution proceeded smoothly to afford (R)-HP(S)PhOMe in 38% yield with 91% ee. A competitive experiment showed that such a dual "soft-soft" interaction was indispensable in the present reaction as the first "soft-soft" interaction activated the phosphinothioates to enable the facile deprotonation and the second "soft-soft" interaction facilitated the addition. Interestingly, both 1H and 31P NMR studies offered some proof for the dual "soft-soft" interaction. Finally, the transformation of the product afforded a cyclic diphosphinate while the reaction of its diastereoisomer delivered a cyclic mono-phosphinate. The recovered (R)-O-methyl phenylphosphinothioate was smoothly converted to various P-chiral compounds without significantly eroded ee. Expanding the application of the dual "soft-soft" interaction in transition metal-catalysis is currently on going in our laboratory.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 22271302), the Science and Technology Commission of Shanghai Municipality (Nos. 20JC1417100 and 21XD1424800), CAS Key Laboratory of Synthetic Chemistry of Natural Substances, and Shanghai Institute of Organic Chemistry.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109294.
References
[1]
A. Mucha, P. Kafarski, L. Berlicki, J. Med. Chem. 54 (2011) 5955–5980.
doi: 10.1021/jm200587f
Uttam Pandurang Patil
. Porous carbon catalysis in sustainable synthesis of functional heterocycles: An overview. Chinese Chemical Letters,
2024, 35(8): 109472-.
doi: 10.1016/j.cclet.2023.109472
Scheme 1. Catalytic asymmetric synthesis of P-chiral phosphinates via KR or DyKAT and our working hypothesis.
Scheme 2. Substrate scope I of the catalytic asymmetric reaction. 1: 0.2 mmol, 2: 0.6 mmol. Isolated yields. ee of the major diastereoisomer was determined by chiral-stationary-phase HPLC analysis. dr was determined by 1H NMR analysis of crude mixture. dr in parenthesis was determined by 1H NMR analysis of an isolated sample. a Gram-scale reaction. b 10 mol% copper(Ⅰ) catalyst used. c 10 mol% Cu(CH3CN)4PF6 and 10 mol% (R)-DTBM-SEGPHOS used at room temperature.
Scheme 3. Substrate scope II of the catalytic asymmetric reaction. 1: 0.2 mmol, 2: 0.6 mmol. Isolated yields. ee of the major diastereoisomer was determined by chiral-stationary-phase HPLC analysis. dr was determined by 1H NMR analysis of crude mixture (in parenthesis of an isolated sample). a Gram-scale reaction. b 10 mol% catalyst loading. c 24 h.
Scheme 4. Control experiments on kinetic resolution of 2a.
Scheme 5. Competitive experiment (1H NMR yields based on 1a and 6 are given).
Figure 1. 31P NMR studies on some mixtures.
Scheme 6. Stereodivergent synthesis to all four stereoisomers with vicinal P-chiral and C-chiral centers.
Scheme 7. Transformations of 3aa and 4aa. Conditions: (a) Cp2ZrHCl, toluene, -78 ℃ to r.t. (b) NaBH4, MeOH, r.t. (c) mCPBA, DCM, 0 ℃ to r.t.

 Login In
Login In




 DownLoad:
DownLoad:

 DownLoad:
DownLoad: