State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
b.
School of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, China
c.
Department of Chemistry, Tsinghua University, Beijing 100084, China
The preparation of Pd-based catalysts with rich electrons and a high atom dispersion rate is of great significance for improving the reactivity of cross-coupling reactions, which is a powerful tool for pharmaceutical and fine chemical synthesis. Here, we report a PdNi single-atom alloy (SAA) catalyst in which isolated Pd single atoms are anchored onto the surface of Ni nanoparticles (NPs) applied for Suzuki coupling reactions and Heck coupling reactions. The 0.1% PdNi SAA exhibits extraordinary catalytic activity (reaction rate: 17,032.25 mmol h−1 gPd−1) toward the Suzuki cross-coupling reaction between 4-bromoanisole and phenylboronic acid at 80℃ for 1 h. The excellent activity is supposed to attribute to the 100 percent utilization rate of Pd atoms and the highly stable surface zero-valance Pd atoms, which provides abundant sites and electrons for the adsorption and fracture of the C-X (X = Cl, Br, I) bond. Moreover, our work demonstrates the excellent application prospect of SAAs for cross-coupling reactions.
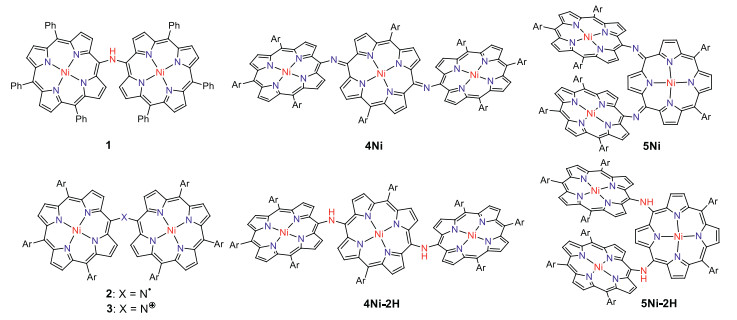
Porphyrin arrays are organic functional molecules with large π-conjugated systems and have potential applications in optoelectronic devices [1-11], sensors [12-15] and photodynamic therapy (PDT) [16-18]. In the last decade, porphyrin arrays with alkynes [19, 20], benzene [21] or heterocycles (such as thiophene [22], pyridine [23], pyrrole [24, 25]) as bridging units have been intensively studied. Porphyrin dimers with a single carbon or heteroatom bridging unit have received much attention due to their unique photophysical properties, chemical properties, and characteristic electronic delocalization [26-37]. In 2006, Arnold et al. reported the first isolation of meso-meso nitrogen-bridged diporphyrinylamine 1, which showed a broadened Soret band and red shift Q bands, indicating substantial electronic interaction between the porphyrins [27]. Ruppert et al. reported meso-meso, β-meso, β-β-nitrogen-bridged diporphyrinylamines [29], which were all synthesized by Buchwald-Hartwig amination. Later, Osuka et al. reported that meso-meso nitrogen-bridged Ni(Ⅱ) porphyrin dimer was cleanly converted into aminyl radical 2 and nitrenium cation 3 by oxidation with PbO2 and tris(4-bromophenyl)aminiumyl hexachloroantimonate (Magic Blue), respectively (Fig. 1) [34]. As an extension, we report here the synthesis of nitrogen-atom bridged Ni(Ⅱ) porphyrin trimers.
Figure 1
Figure 1.N-Bridged porphyrin oligomers. Ar = 3, 5-di-tert-butylphenyl.
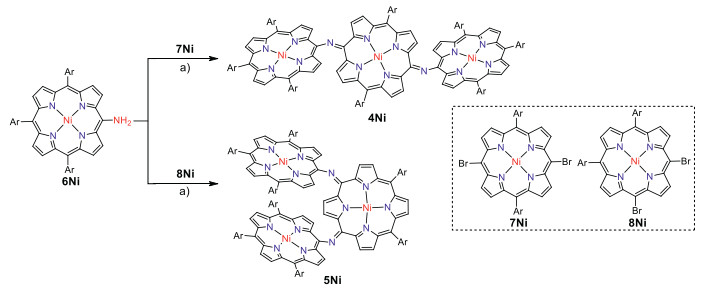
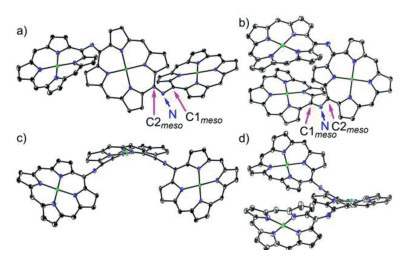
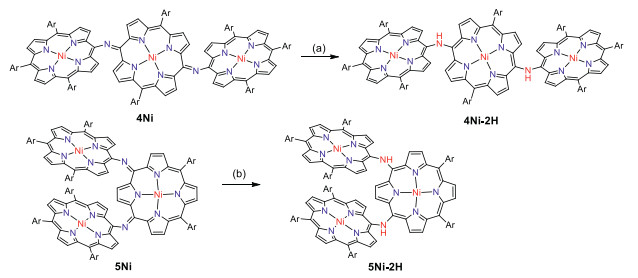
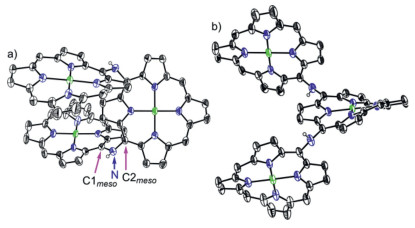
First we attempted to synthesize linear NH-bridged porphyrin trimer 4Ni-2H by the similar Buchwald-Hartwig amination of 5, 15-dibromo Ni(Ⅱ)porphyrin 7Ni with 5-amino Ni(Ⅱ)porphyrin 6Ni [34]. A 4:1 solution of 6Ni and 7Ni in toluene was heated at 100 ℃ for 12 h in the presence of 0.4 equiv. Pd(OAc)2, 0.4 equiv. BINAP, and 7 equiv. t-BuOK (Scheme 1). To our surprise, only a linear trimer 4Ni bearing a central quinodiimine-type porphyrinoid unit was obtained in 38% yield. The matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrum showed the parent ion of 4Ni at m/z 2627.3453 [M]+ (calcd. for (C172H192N14Ni3)+ = 2627.3509) (Fig. S13 in Supporting information), which is smaller by two than the expected parent ion peak of 4Ni-2H. The structure of 4Ni has been revealed by X-ray diffraction structural analysis (Fig. 2 and Fig. S17 in Supporting information). The bond lengths of C2meso-N (1.300(6) Å and 1.304(6) Å) are distinctly shorter than those of C1meso-N (1.412(6) Å and 1.395(7) Å). The 1H NMR spectrum of 4Ni showed broadened signals at room temperature in CDCl3 (Fig. S3 in Supporting information), which gradually changed to sharp peaks upon cooling down to −60 ℃ (Fig. S4 in Supporting information) [38], suggesting conformational motions at room temperature, which are comparable or faster than 1H NMR timescale. It is noteworthy that four doublets due to the b-protons of the central quinodiimine unit were observed in the up-field shifted region at 7.77, 6.76, 5.70 and 3.99 ppm.
Scheme 1
Scheme 1.
Syntheses of meso-meso N-bridged porphyrinoid trimers. Conditions: a) Pd(OAc)2, BINAP, t-BuOK, toluene, 100 ℃, 12 h. Ar = 3, 5-di-tert-butylphenyl.
Figure 2.
X-ray single crystal structure of 4Ni and 5Ni. (a) Top view and (b) side view of 4Ni, (c) top view and d) side view of 5Ni. The thermal ellipsoids are on 30% probability level. Solvent molecules, 3, 5-di-tert-butylphenyl groups, and hydrogens are omitted for clarity.
Similarly, Buchwald-Hartwig amination of 5, 10-dibromo Ni(Ⅱ)porphyrin 8Ni with 6Ni afforded l-shaped bent trimer 5Ni in 25% yield. The quinodiimine structure of 5Ni has been also confirmed by X-ray analysis. 5Ni shows that the bond lengths of C2meso-N (1.299(5) Å and 1.302(6) Å) are shorter than those of C1meso-N bonds (1.399(5) Å and 1.413(6) Å) (Fig. 2 and Fig. S18 in Supporting information). The 1H NMR spectrum of 5Ni showed broadened signals at room temperature that became sharp and complicated signals at −60 ℃ in CDCl3 (Figs. S5 and S6 in Supporting information). In line with the quinodiimine structure, the corresponding β-protons were observed in the high field at 7.07, 6.73, 6.42, 6.33, 5.66, 4.33, and 3.74 ppm.
The structural data of 4Ni shows that lengths of C1meso-N bonds (1.412(6) Å and 1.395(7) Å) bond to the terminal porphyrin units are longer than C2meso-N (1.300(6) Å and 1.304(6) Å) attached to the central quinodiimine units. Similarly, 5Ni shows that lengths of C1meso-N bonds (1.399(5) Å and 1.413(6) Å) bond to the terminal porphyrin units are longer than C2meso-N (1.299(5) Å and 1.302(6) Å) attached to the central quinodiimine units. The observed short C2meso-N bond lengths in 4Ni and 5Ni indicated its double bond characters significantly [34], which further proved the structure of 4Ni and 5Ni to be N-bridged (rather than NH-bridged) porphyrin trimer. The dihedral angles between the terminal porphyrins and terminal porphyrin, terminal porphyrin and central quinodiimine are 66.81(3)°, 56.34(3)° and 58.06(3)° in 4Ni, and 6.83(3)°, 42.67(3)° and 39.34(3)° in 5Ni (Fig. 2 and Figs. S17 and S18 in Supporting information).
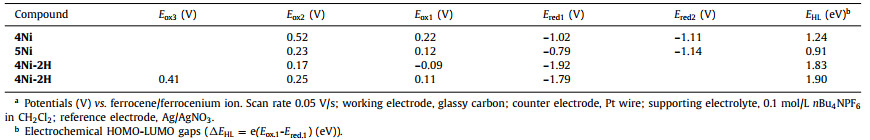
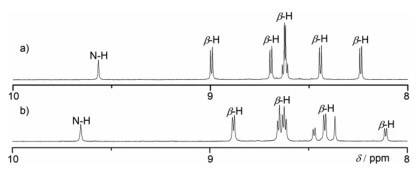
Electrochemical properties of 4Ni and 5Ni were examined by cyclic voltammetry and differential-pulse voltammetry in CH2Cl2 against a ferrocene/ferrocenium ion couple (Table 1 and Table S4 in Supporting information). Reversible oxidation waves were recorded at 0.22 and 0.52 V for 4Ni, and at 0.12 and 0.23 V for 5Ni. Reversible reduction waves were observed at −1.02 and −1.11 V for 4Ni, and at −0.79 and −1.14 V for 5Ni (Figs. S20 and S21 in Supporting information). As a result, the electrochemical HOMO-LUMO gaps of 4Ni and 5Ni were determined to be 1.24 and 0.91 eV, respectively. The observed reversible reduction waves of 4Ni and 5Ni encouraged us to examine their chemical reduction. After many attempts, we found that reduction of 5Ni with aqueous hydrazine in CH2Cl2 afforded 5Ni-2H quantitatively (Scheme 2). Curiously, 4Ni was not reduced with aqueous hydrazine but was reduced quantitatively to give 4Ni-2H with NaBH4 and Pd/C in CH2Cl2/CH3OH. 1H NMR spectra of both 4Ni-2H and 5Ni-2H are very simple, reflecting their symmetric structures with signals of the β-protons appearing in the range of 8–9 ppm (Fig. 3 and Figs. S7 and S8 in Supporting information). The structure of 5Ni-2H has been confirmed by single crystal X-ray diffraction analysis (Fig. 4 and Fig. S19 in Supporting information). In 5Ni-2H, the bond lengths of the C2meso-N bond and the C1meso-N bond are similar, being 1.409(8) Å, 1.406(8) Å and 1.393(7) Å, 1.434(11) Å, respectively, in line with the assigned structures. In addition, the dihedral angles between the terminal porphyrins and the central porphyrin are 58.29(7)° and 58.15(7)°, which are larger than those on 5Ni (42.67(3)° and 39.34(3)°).
Table 1
Table 1.
Electrochemical measurement of 4Ni, 5Ni, 4Ni-2H and 5Ni-2H performed in CH2Cl2 at room temperature.a
Figure 4.
X-ray single crystal structure of 5Ni-2H. (a) Top view and (b) side view. The thermal ellipsoids are on 30% probability level. Solvent molecules, 3, 5-di-tert-butylphenyl groups, and hydrogens except those connected to N atoms are omitted for clarity.
The unexpected formation of 4Ni and 5Ni may be ascribed to the facile oxidation of 4Ni-2H and 5Ni-2H under the amination reaction conditions. These trimers have the central electron-rich Ni(Ⅱ) porphyrin bearing 5, 15 or 5, 10-aminoporphyrin units. Thus, we examined the electrochemical properties of 4Ni-2H and 5Ni-2H (Table 1 and Table S4 in Supporting information). Actually, the reversible oxidation waves were observed at −0.09 and 0.17 V for 4Ni-2H, and at 0.11, 0.25 and 0.41 V for 5Ni-2H (Figs. S22 and S23 in Supporting information). It is thus conceivable that 4Ni-2H and 5Ni-2H are oxidized under the amination conditions with air. So, when we try to oxidized them with PbO2 and Magic Blue, neither aminyl radical nor nitrenium cation was found. The possible reason may be that the quinodiimine unit is more stable than other species.
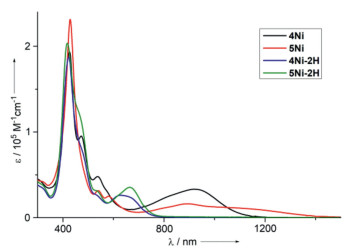
The UV–vis-NIR absorption spectra of 4Ni, 5Ni, 4Ni-2H and 5Ni-2H in CH2Cl2 are shown in Fig. 5. 4Ni shows two split Soret bands at 426 and 472 nm, a Q-band at 537 nm, and a broadened Q-like band at 915 nm. 5Ni shows a Soret band at 429 nm, Q-bands at 540 and 581 nm, and a broadened Q-like band at 892 nm. Both 4Ni and 5Ni exhibit characteristic absorption spectra of quinonoidal porphyrinoid arrays [39-42]. 4Ni-2H shows a Soret band at 423 nm, and a Q-band at 627 nm. Similarly to 4Ni-2H, the absorption spectrum of 5Ni-2H shows a Soret band at 418 nm, and a Q-band at 664 nm. In particular, 4Ni and 5Ni display the lowest energy band reaching to 1200 nm and 1400 nm, respectively.
Figure 5
Figure 5.
UV–vis-NIR absorption spectra of 4Ni (black line), 5Ni (red line), 4Ni-2H (blue line) and 5Ni-2H (green line) in CH2Cl2.
Density functional theory (DFT) calculations clearly indicated that both the HOMO of 4Ni and HOMO-1 5Ni were localized at terminal porphyrin units, whereas both LUMOs of 4Ni and 5Ni were localized at the central quinodiimine units (Figs. S28 and S29 in Supporting information). Time-dependent density functional theory (TD-DFT) calculations indicated that the absorption bands around 1000 nm of trimers 4Ni and 5Ni resulted from the transition from HOMO to LUMO of 4Ni and HOMO-1 to LUMO of 5Ni, respectively (Figs. S24 and S25 in Supporting information). These results show that both absorption bands around 1000 nm of 4Ni and 5Ni could be assigned to charge transfer (CT) band.
In summary, we synthesized N-bridged porphyrinoid trimers 4Ni and 5Ni having the central quinodiimine through Buchwald-Hartwig amination, under which the oxidations of the NH-bridged porphyrin trimers 4Ni-2H and 5Ni-2H proceeded smoothly. The trimer 4Ni-2H was obtained by reduction with NaBH4 and Pd/C, while 5Ni-2H was obtained by reduction with aqueous hydrazine. The structures of 4Ni, 5Ni and 5Ni-2H were determined by X-ray diffraction analysis. The UV–vis-NIR absorption spectra showed that the trimers 4Ni and 5Ni have the lowest energy band reaching to 1200 nm and 1400 nm, respectively. These N-bridged porphyrinoid trimers exhibited interesting spectral properties. Further exploration of cyclic or larger N-bridged porphyrinoid arrays is ongoing in our laboratory.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The work at Hunan Normal University was supported by the National Natural Science Foundation of China (Nos. 21772036, 22071052, 21602058, 21702057), the Science and Technology Planning Project of Hunan Province (No. 2018TP1017), and the Scientific Research Fund of Hunan Provincial Education Department (No. 19A331), and Hunan Provincial Innovation Foundation for Postgraduate (No. CX20210473).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.061.
References
[1]
N. Miyaura, A. Suzuki, Chem. Rev. 95 (1995) 2457–2483.
doi: 10.1021/cr00039a007
[2]
A. Fihri, M. Bouhrara, B. Nekoueishahraki, et al., Chem. Soc. Rev. 40 (2011) 5181–5203.
doi: 10.1039/c1cs15079k
[3]
L. Yu, Y. Huang, Z. Wei, et al., J. Org. Chem. 80 (2015) 8677–8683.
doi: 10.1021/acs.joc.5b01358
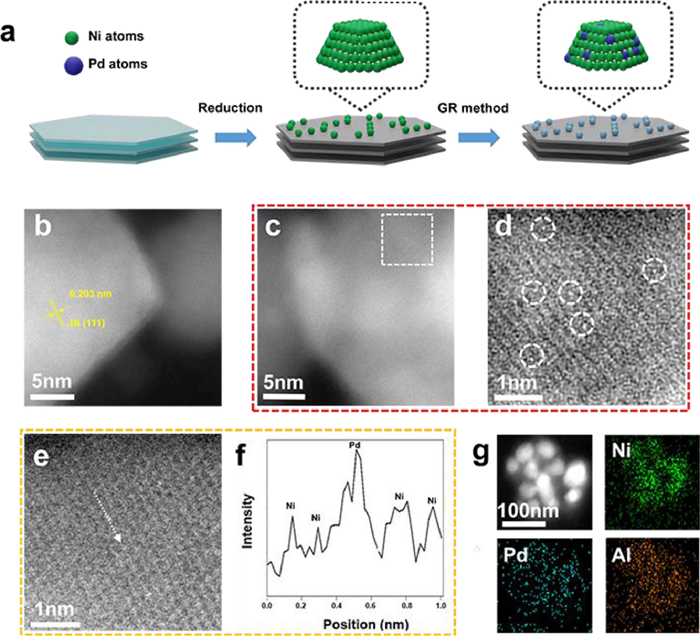
Figure 1. Synthesis process and morphological characterizations of 0.1% PdNi SAA. (a) Schematic illustration of the synthesis approach of bimetallic PdNi samples. (b, c) AC-HAADF-STEM images of 0.1% PdNi SAA. The crystal plane spacing of 0.203 nm indexed to the (111) plane of the metallic Ni. (d) The enlarged area labeled by the white dashed line of (c). (e) enlarged STEM image and (f) corresponding intensity profile of 0.1% PdNi SAA. (g) EDS mapping images of 0.1% PdNi SAA.
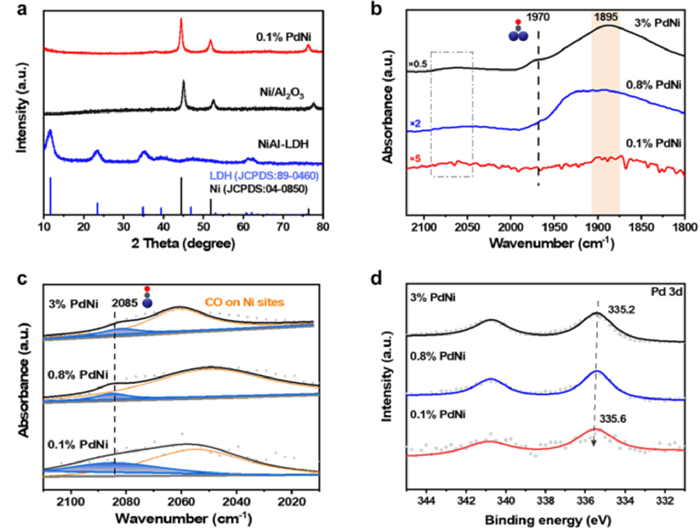
Figure 2. Characterizations of catalyst structure. (a) XRD patterns of NiAl-LDH, Ni/Al2O3, and 0.1% PdNi SAA. (b) CO-DRIFTS spectra of 0.1% PdNi SAA, 0.8% PdNi, and 3% PdNi samples, within 2120–1800 cm−1 by flowing He gas for 20 min. (c) The enlarged and peak fitting curves of (b) within 2120–2010 cm−1. (d) Pd 3d XPS spectra of 0.1% PdNi SAA, 0.8% PdNi, and 3% PdNi samples.
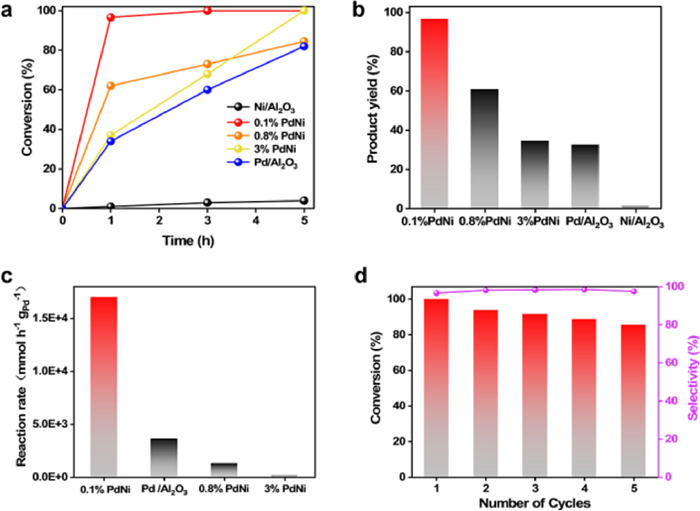
Figure 3. Catalytic performance of the catalysts in Suzuki coupling reaction. (a) Conversion of 4-bromoanisole in Suzuki coupling reaction versus time over 0.1% PdNi SAA, 0.8% PdNi, 3% PdNi, Pd/Al2O3, and Ni/Al2O3. (b) Yields of the Suzuki coupling reaction of 4-methoxybiphenyl over 0.1% PdNi SAA, 0.8% PdNi, 3% PdNi, Pd/Al2O3, and Ni/Al2O3 after 1 h. (c) The reaction rate of 0.1% PdNi SAA, 0.8% PdNi, 3% PdNi, and Pd/Al2O3. (d) Recyclability and stability of 0.1% PdNi SAA. Reaction conditions: deionized water (5 mL), ethanol (5 mL), 0.1% PdNi SAA catalysts (20 mg), 4-bromoanisole (0.33 mmol), phenylboronic acid (0.66 mmol), and K2CO3 (0.5 mmol) at 80℃.
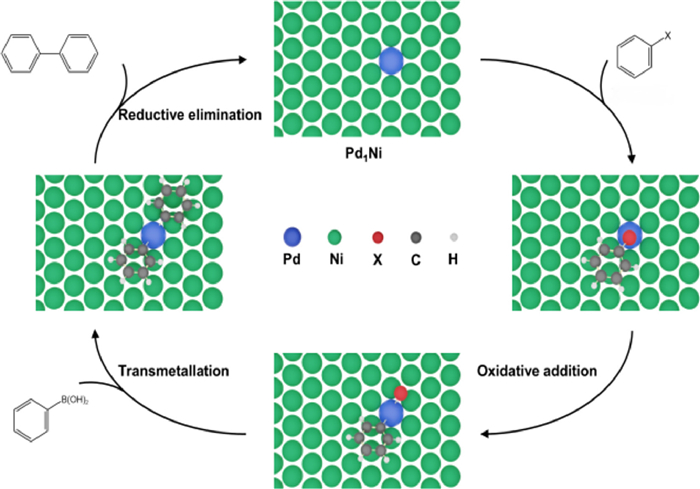
Figure 4. Proposed mechanism. Proposed reaction paths for the Suzuki cross-coupling process over 0.1% PdNi SAA.

 Login In
Login In




 DownLoad:
DownLoad:

 DownLoad:
DownLoad: