Citation:
Yongjie Li, Mingjie Huang, Wen-Da Oh, Xiaohui Wu, Tao Zhou. Efficient activation of sulfite for reductive-oxidative degradation of chloramphenicol by carbon-supported cobalt ferrite catalysts[J]. Chinese Chemical Letters,
;2023, 34(10): 108247.
doi:
10.1016/j.cclet.2023.108247
Efficient activation of sulfite for reductive-oxidative degradation of chloramphenicol by carbon-supported cobalt ferrite catalysts
Activation of (bi)sulfite (S(Ⅳ)) by metal oxides is strongly limited by low electrons utilization. In this study, two carbon-supported cobalt ferrites spinels (CoFe2O4 QDs-GO and CoFe2O4 MOFs-CNTs) have been successfully synthesized by one-step solvothermal method. It was found that both catalysts could efficiently activate S(Ⅳ), with rapid reductive dechlorination and then oxidative degradation of a recalcitrant antibiotic chloramphenicol (CAP). Characterizations revealed that CoFe2O4 spinels were tightly coated on the carbon bases (GO and CNTs), with effectiveness of the internal transfer of electrons. O2˙− was identified for the reductive dechlorination of CAP, with simultaneously detection of both •OH and SO4˙− responsible for further oxidative degradation. The sulfur oxygen radical conversion reactions and molecular oxygen activation would occur together upon the carbon-based spinels. Spatial-separated interfacial reductive-oxidation of CAP would occur with dechlorination of CAP by O2˙− on the carbon bases, and oxidative degradation of intermediates by SO4˙−/•OH upon the CoFe2O4 catalysts.
Lanthanide complexes are crystalline hybrid materials assembled by trivalent lanthanide (Ln3+) ions and organic ligands [1-3]. Benefiting from the antenna effect, characteristic luminescence of Ln3+ ions, especially Eu3+ and Tb3+ ions, are well sensitized by the light-harvesting ligands via a series of energy transfer processes [4-6]. These complicated photoluminescence pathways determine the sensitive nature to circumstance details, thereby providing an effective platform for chemically responding to metal cations, oxoanions, organic solvents, and biomolecules [7-10]. However, it is commonly believed that luminescent lanthanide complexes are unsuitable for water-bearing systems, because the stretching vibration of hydroxyl groups in water molecules confines the energy transfer process from the triplet state of ligand to the receiving energy level of Ln3+ ions, leading to dramatic luminescence quenching [11-13]. This greatly limits the practicality of lanthanide-based luminescence complexes. Therefore, it is meaningful, though challengeable, to develop a water-stable even water-improving luminescence lanthanide complex.
Enhancing hydrophobicity of luminophores and thereafter inhibiting the immediate contact with solvent water molecules is a potential mean to settle the above-mentioned water-induced luminescence quenching issue [14]. Based on this consideration, (HNEt3)+[Eu(DBM)4]– (briefly as Eu-DBM, DBM = dibenzoylmethane) is selected as a luminophore (Figs. S1 and S2 in Supporting information) [15]. Firstly, its phenyl-surrounding outside surface provides strong hydrophobicity. Secondly, it has coordination-saturated lanthanide centre, which could decrease the impact of coordination solvent molecular replacement and further weaken the water-induced effect to lanthanide luminescence. Thirdly, its anionic framework is helpful to the dispersibility in polar solvents such as water. Therefore, in this work, the Eu-DBM material is selected as a luminophore to study its potential on water-induced luminescence improvement. For further enhancing the dispersibility and stability, the nanosheet-like Eu-DBM material is also prepared. Then, its application on sensitive detection toward extended organic solvents in water has been investigated.
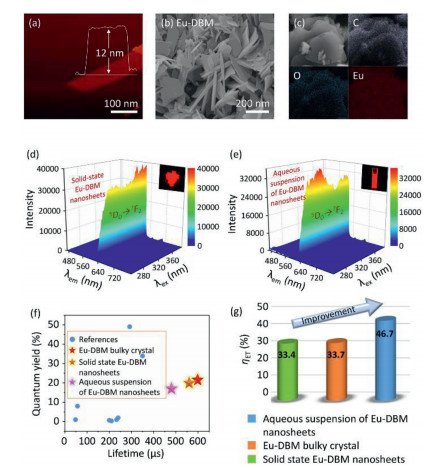
A top-down strategy was utilized to prepare the Eu-DBM nanosheet by two facile steps. The bulky crystal of Eu-DBM was firstly prepared in terms of a previous method [15]. It was then exfoliated into nanosheets under 720 W ultrasonication for 24 h at room temperature. Atomic force microscopy (AFM) image revealed its two-dimensional (2D) sheet-like morphology with a height of uniform 12 nm (Fig. 1a). Scanning electron microscopy (SEM) image further confirmed a sheet-like appearance with the size of 100–200 nm (Fig. 1b). Fourier transition infrared (FT-IR), ultraviolet-visible absorption (UV-vis) thermogravimetric differential thermal analysis (TG-DTA) spectra, and the powder X-ray diffraction pattern of the Eu-DBM nanosheet held similar patterns with those of bulky ones reported previously (Figs. S3–S6 in Supporting information) [15]. Furthermore, elemental mapping analysis further proved the composition consisted of C, O and Eu elements (Fig. 1c). Elementary analysis, X-ray photoelectron spectroscopy (XPS), and energy disperse spectroscopy (EDS) presented an agreeable proportion of elements with crystallographic data (Table S1 in Supporting information) [15]. These results indicated that the obtained Eu-DBM nanosheets had the same structure and composition as their bulky crystals.
Figure 1
Figure 1.
AFM (a), SEM (b), and elemental mapping (c) images of Eu-DBM nanosheets. 3D photoluminescent spectra of Eu-DBM nanosheet in solid state (d) and in aqueous solution (e). (f) Comparison of lifetime and quantum yield in this work with previous reports. (g) Comparison of Eu-DBM bulky crystal and nanosheet in solid state and in aqueous suspension.
The three-dimensional (3D) photoluminescence (PL) spectra of Eu-DBM nanosheet exhibited strong emission at 614 nm throughout the excitation from 250 nm to 390 nm, rendering stable and intense red-light emission with the Commission Internationale de L'Eclairage (CIE) coordination of (0.648, 0.330) (Fig. 1d and Fig. S7 in Supporting information). This excitation-wavelength independence in such a broad range was rarely reported in the luminescence lanthanide complexes [12, 13]. The unsplit band at 614 nm implied a highly symmetric structure of the Eu-DBM complex [16]. The electric dipole transition (5D0→7F2) at 614 nm was approximately 22-fold stronger than the magnetic one (5D0→7F1) at 592 nm, indicating the highly symmetric coordination environment of central Eu3+ ion in the complex [17]. Noteworthily, after dispersing the Eu-DBM nanosheet in water, the 1 g/L suspension presented the nearly same 3D PL contour as the solid-state one, exhibiting the strong red-light emission (Fig. 1e). The time-dependent curve showed that the luminescent intensity at 614 nm could remain at least 24 h with a < 5% declination (Fig. S8 in Supporting information). This excellent stability originated from the good dispersibility of the complex nanosheets in water.
The presence of strong Eu3+ characteristic luminescence and the absence of DBM ligand fluorescence suggested an effective antenna effect in the Eu-DBM complex: the adsorbed UV light by DBM ligand could effectively transfer to the Eu3+ ion, and then sensitize its characteristic luminescence. To confirm this hypothesis, the sensitization efficiency (ηET) was subsequently calculated in terms of the following equations Eqs. 1–3 [18-20].
(1)
(2)
(3)
According to Eq. 1, Φoverall was the overall luminescence quantum yield measured by the integrated sphere method (Table S2 in Supporting information). ΦLn was the intrinsic quantum yield of the lanthanide luminescence, obtained in terms of Eq. 2, where the observed lifetime (τobs) was determined by monitoring the emission decaying curve within the 5D0→7F2 transition at 614 nm (Figs. S9–S11 in Supporting information). The calculated radiative lifetime (τrad) could be calculated through Eq. 3, where AMD, 0 was the spontaneous emission probability of the magnetic dipole transition and equated to 14.65 s−1 for Eu3+ ion (5D0→7F1); n represented the refractive index of the tested sample (n equated to 1.55 for solid-state Eu-DBM and 1.33 for Eu-DBM nanosheets aqueous suspension); Itot and IMD were the integrated emission of the total 5D0→7FJ transition and the 5D0→7F1 transition, respectively. As a result, the Eu-DBM bulky crystal and nanosheet in solid state and in aqueous suspension exhibited high τobs and Φoverall values, surpassing most of the reported luminescent lanthanide complexes and nearly all the Eu-DBM-based complexes (Fig. 1f). Compared with the solid-state Eu-DBM, the 1 g/L aqueous suspension of Eu-DBM nanosheets showed only slight declination on τobs and Φoverall (Table S2 and Figs. S9–S11). Noteworthily, the ηET in Eu-DBM aqueous suspension was calculated as 46.7%, a 13.0% and 13.3% improvement compared with that of the bulky crystal and solid-state nanosheets (Fig. 1g). This improvement on sensitization efficiency states the increasing energy transfer from the DBM ligand to the central Eu3+ ions. In other words, the water solvent improved the luminescence of Eu3+ ion, realizing the water-induced luminescence improvement which is rarely reported previously (Table S2).
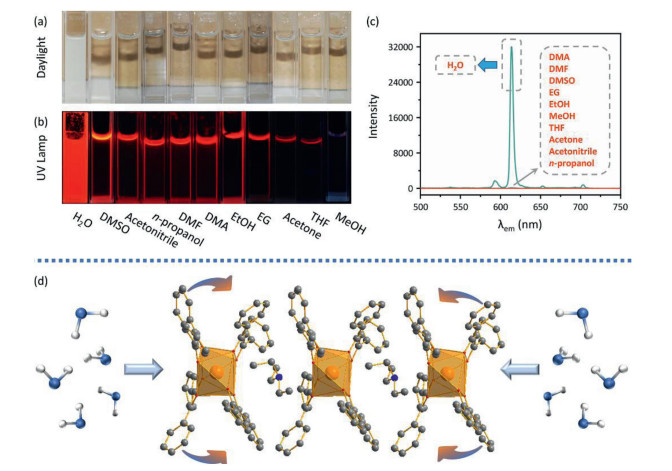
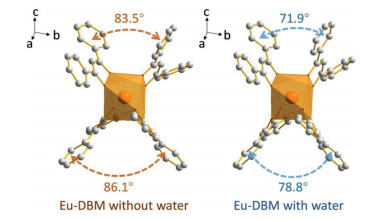
The mechanism of this water-induced luminescence improvement in the Eu-DBM complex was then explored. Firstly, the Eu-DBM was dispersed in various common solvents to prepare 1 g/L solution or suspension. As seen in Fig. 2a, the complex exhibited poor solubility in water but good in nearly all other common organic solvents. This derived from the strong hydrophobic property due to the phenyl-surrounding outside surface of Eu-DBM. Comparatively, the aqueous suspension of the Eu-DBM nanosheets presented strong and uniform red-light-emission of characteristic Eu3+ ion (Figs. 2b and c). However, the Eu-DBM solution in various common organic solvents was obviously quenched. These excellent and unorthodox photophysical properties of Eu-DBM in water system could attribute to the insolubility and aggregation state which avoided the immediate contact between water molecules and the central Eu3+ ions. Secondly, given that water possesses the highest surface tension and polarity among all solvents, it could suppress surficial phenyl groups and yield structural modification. According to Fig. S5, the PXRD pattern of the Eu-DBM nanosheet had strong peak at 7.0o, which indicated that the (200) plane was exposed to water solvent, as shown in Fig. 2d. The strong surface tension of solvent water molecules could compress the hydrophobic phenyl groups inward, resulting in the more planarity between the neighboring phenyl and carbonyl groups. This enhanced the π-π conjugation and suppressed the stretching vibration of the C–C bond of benzene rings. After the benzene rings are fixed, the energy loss caused by the vibration of the chemical bonds were reduced, making the more effective sensitization of Eu3+ ions (Fig. 2d). Moreover, the dihedral angle and distances between neighboring phenyl/carboxyl planes was decreasing after putting Eu-DBM nanosheets into water environment, which promoted the through-space charge transfer (TSCT) process [21-23]. To prove this hypothesis, the Eu-DBM molecule in the water environment was geometrically optimized by theoretical calculation by using Material Studio 8.0 software with GGA/BLYP basis set [24]. As shown in Fig. 3, under the effect of the water molecule, a more planarity between phenyl and carbonyl groups could be observed. The dihedral angle between the neighboring phenyl/carbonyl planes were also reduced by 11.6° and 7.3°, respectively, compared with the original structure. This confirmed that the water-induced luminescence improvement primarily resulted from the TSCT process as discussed above.
Figure 2
Figure 2.
Photo images of Eu-DBM samples in various solvents under daylight (a) and UV lamp (b). Photoluminescent spectra of Eu-DBM samples in various solvents upon the excitation of 304 nm (c). Mechanism of water-induced luminescence improvement (d).
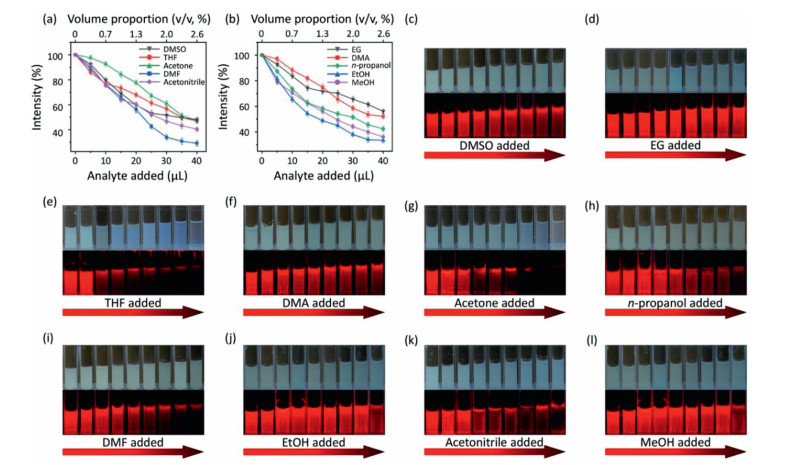
Considering that the as-prepared Eu-DBM nanosheet presented excellent luminescence improvement in water and obvious quenching in other organic solvents, it could act as a luminescence sensor toward extensive organic solvents in water, being an indicator to monitor water purity. Based on this, various organic solvents were gradually titrated into 1 g/L Eu-DBM aqueous suspension. By monitoring their luminescence intensity at 614 nm upon the 304 nm excitation, the relationship between volume proportion (v/v) and quenching efficiency was depicted in Figs. 4a and b. The luminescence intensity dramatically decreased upon the addition of various organic solvents. When the volume proportion of added organic solvent was approximate 2.5%, the luminescence intensity dropped by about 50%. In other words, the nanosheets exhibited similar and sensitive changes in luminescence intensity toward nearly all organic solvents. To the best of our knowledge, this colligative property toward such extensive sensibility was rarely reported and applicable for water purity monitor. As shown in Figs. 4c–l, the Eu-DBM aqueous suspension turned to be more pellucid as the gradual addition of various organic solvents. The luminescent intensity observed by naked eyes also presented an obvious decrease under the 365 nm UV lamp.
Figure 4
Figure 4.
(a, b) Relationship between the relative luminescent intensity of Eu-DBM sensor and the added quantity of organic solvents. Photo images under daylight and UV lamp of Eu-DBM sensor upon adding DMSO (c), EG(d), THF (e), DMA (f), acetone (g), n-propanol (h), DMF (i), EtOH (j), acetonitrile (k) and MeOH (l).
Then, we quantitatively examined the luminescent quenching coefficient (KSV) of the Eu-DBM sensor toward various organic solvents in water via the distinguished Stern-Volmer (SV) equation: I0/I – 1 = KSV × [C]n, where I0 and I were the luminescent intensity before and after adding analytes, and [C] was the molar concentration of the analyte [25, 26]. The parameter n equalled to 1 when the SV curve displayed a linear relationship. According to Figs. S12–S31(Supporting information), the SV curve of the Eu-DBM sensor displayed a linear relationship in the concentration range of 0–0.6 mol/L toward DMA, DMF, DMSO, EtOH, MeOH, acetonitrile, and n-propanol, but a distinct curvature toward THF, acetone, and ethylene glycol (EG). The calculated KSV values were in order of acetone (8.85 L/mol) > THF (7.35 L/mol) > DMF (7.22 L/mol) > EtOH (6.54 L/mol) > n-propanol (4.79 L/mol) > acetonitrile (2.54 L/mol) > MeOH (2.52 L/mol) > DMA (2.78 L/mol) > DMSO (2.43 L/mol) > EG (2.11 L/mol). Subsequently, the limitations of detection (LODs) were calculated via the formula of 3σ/KSV, where σ is the standard deviation of the luminescent intensity for five-time blank measurement at 2 min intervals [27-30]. As a result, the LODs toward various organic solvents ranged from 4.07 × 10−4 mol/L to 1.43 × 10−3 mol/L. In order to improve the detection convenience, the relationship between the volume ratio of the 10 organic solvents and the relative luminescence intensity was averaged and the polynomial fitting was performed (Fig. S32 in Supporting information). The relationship between them is approximately fitted as y = 5.23x2 – 35.47x + 100.00 and R2 = 0.999. This extensive sensibility toward nearly all organic solvents in water demonstrated that the as-prepared Eu-DBM nanosheets could be utilized to ratiometrically determine the organic solvents in water. In addition, the sensor could be regenerated and reused for at least five cycles by centrifugation of the solution after use and washing several times with water (Figs. S33–S42 in Supporting information).
In conclusion, the water-stable Eu-DBM nanosheets exhibited exceptional water-induced luminescence improvement phenomenon, with a 13% improvement for sensitization efficiency. This attributes to the combined effect of hydrophobic-caused aggregation and through-space charge transfer in the water environment. Moreover, the well-dispersed aqueous suspension of Eu-DBM nanosheets could act as a luminescence sensor which responses to trace organic solvents in water, with good universality and recyclability. In this work, the water-induced luminescence improvement and extensive sensibility toward organic solvents in water were rarely reported previously in lanthanide-based materials. The successful development of this material provided a new platform for luminescent sensors in the water system, being practical values on monitoring water quality.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank the National Natural Science Foundation of China (No. 22075071), and Reform and Development Fund Project of Local University supported by the Central Government.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.cclet.2021.08.080.
Y. Yuan, Oxidative of organic compounds by Oxysulfur radicals in the presence of transition metal ions and sulfite, Université Clermont Auvergne [2017-2020]; Université de Wuhan (Chine), 2018.
Y. Yuan, Oxidative of organic compounds by Oxysulfur radicals in the presence of transition metal ions and sulfite, Université Clermont Auvergne [2017-2020]; Université de Wuhan (Chine), 2018.
Peng Wang
, Daijie Deng
, Suqin Wu
, Li Xu
. Cobalt-based deep eutectic solvent modified nitrogen-doped carbon catalyst for boosting oxygen reduction reaction in zinc-air batteries. Chinese Journal of Structural Chemistry,
2024, 43(1): 100199-100199.
doi: 10.1016/j.cjsc.2023.100199
Xiaoling WANG
, Hongwu ZHANG
, Daofu LIU
. Synthesis, structure, and magnetic property of a cobalt(Ⅱ) complex based on pyridyl-substituted imino nitroxide radical. Chinese Journal of Inorganic Chemistry,
2025, 41(2): 407-412.
doi: 10.11862/CJIC.20240214
[20]
Yanhua Peng
, Xin Yu
, Ting Wang
. Adaptive nanoconfined Fenton-like reactions: Tailoring carbon pathways for sustainable water treatment and energy harvesting. Chinese Chemical Letters,
2024, 35(12): 110198-.
doi: 10.1016/j.cclet.2024.110198
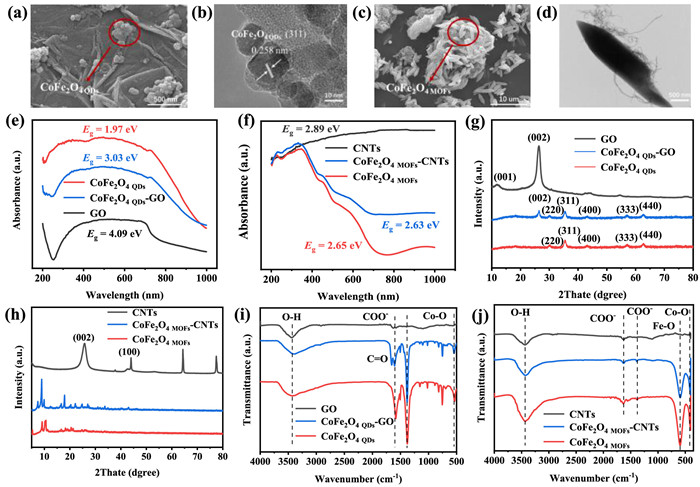
Figure 1. SEM and HR-TEM images of (a, b) CoFe2O4 QDs-GO and (c, d) CoFe2O4 MOFs-CNTs, respectively. UV–vis DRS spectra (e, f), XRD (g, h) and FTIR patterns (i, j) of the two comparative materials groups (CoFe2O4 QDs-GO and CoFe2O4 MOFs-CNTs).
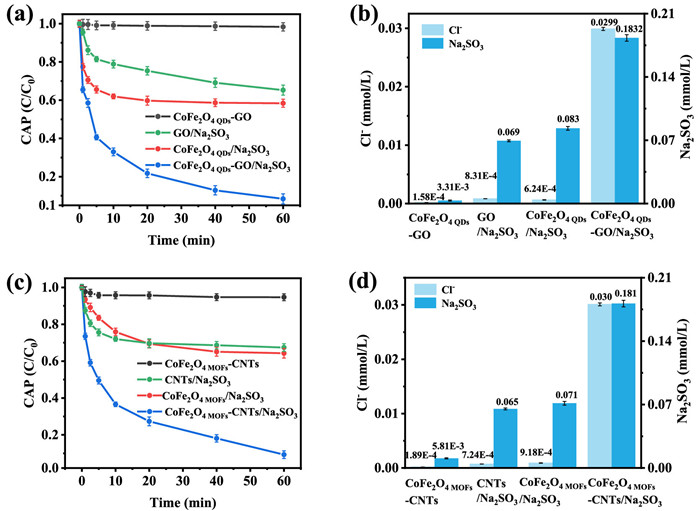
Figure 2. Comparative CAP degradation and the related chloride releases with Na2SO3 consumptions in the comparative systems: (a, b) CoFe2O4 QDs-GO, GO/Na2SO3, CoFe2O4 QDs/Na2SO3, CoFe2O4 QDs-GO/Na2SO3, and (c, d) CoFe2O4 MOFs-CNTs, CNTs/Na2SO3, CoFe2O4 MOFs/Na2SO3, CoFe2O4 MOFs-CNTs/Na2SO3 systems. Initial conditions were: 0.1 g /L catalyst, 0.2 mmol/L Na2SO3, 10 mg/L CAP, pH 9.0 and 25 ℃.
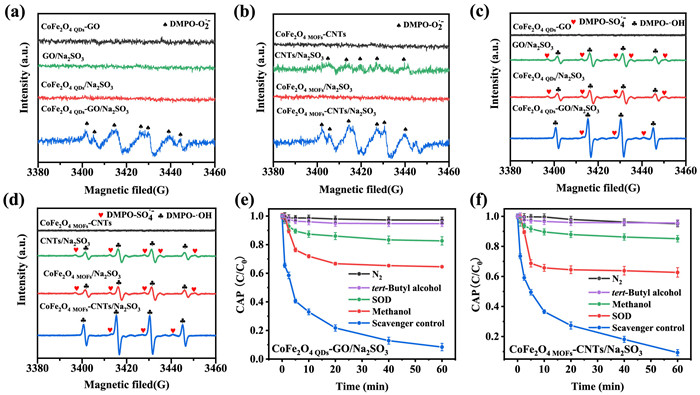
Figure 3. DMPO spin-trapping EPR spectra with (a, b) or without methanol solvent (c, d) in the eight systems (the comparative groups of CoFe2O4 QDs-GO and CoFe2O4 MOFs-CNTs with Na2SO3 or not). (e, f) Effect of N2 sparing, methanol or SOD scavengers in on CAP degradation in the CoFe2O4 QDs-GO and CoFe2O4 MOFs-CNTs systems with Na2SO3. Initial conditions were: 0.1 g /L catalyst, 0.2 mmol/L Na2SO3, 10 mg/L CAP, pH 9.0 and 25 ℃.
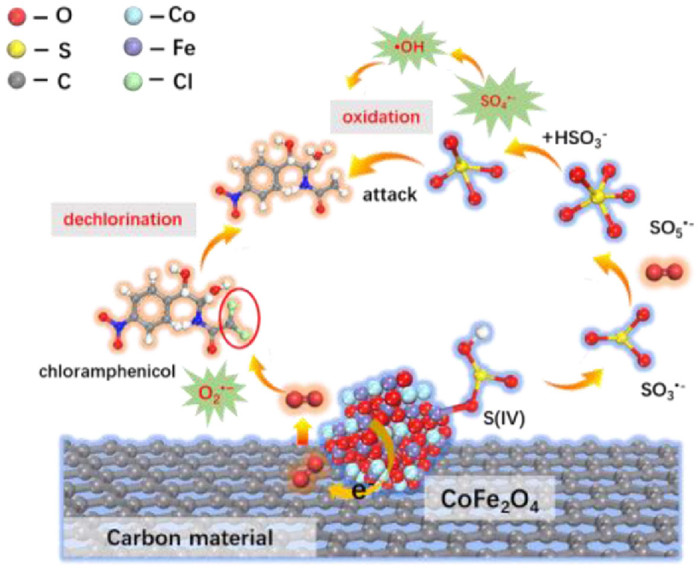
Figure 4. A proposed mechanistic scheme of the spatial-separated reductive-oxidation of CAP in carbon-based Co2FeO4 catalysts/S(Ⅳ) systems.

 Login In
Login In




 DownLoad:
DownLoad:
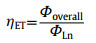
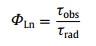
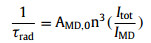

 DownLoad:
DownLoad: