Login In
Login InEnhanced removal of phosphonates from aqueous solution using PMS/UV/hydrated zirconium oxide process
-
* Corresponding author.
E-mail address: bimingliu@tsinghua.edu.cn (B. Liu).
Figures(5)
Citation:
Shunlong Pan, Xi Nie, Xinrui Guo, Hao Hu, Biming Liu, Yongjun Zhang. Enhanced removal of phosphonates from aqueous solution using PMS/UV/hydrated zirconium oxide process[J]. Chinese Chemical Letters,
;2023, 34(4): 107620.
doi:
10.1016/j.cclet.2022.06.043

Figures(5)




Catalytic CO oxidation plays a vital role in solving raised air pollution problems induced by CO emission, which is produced from incomplete combustion of carbon-based compounds in automobiles and industrial processes [1-3]. Searching for a suitable catalyst effectively catalytic oxidation of CO is vital for purpose of achieving removal of CO pollutants. As previous researches reported, noble metal materials such as Au, Ag, Pt, Pd, and Rh are available catalysts in oxidizing CO [4-7]. However, noble metal materials are source scarce and price expensive. Accordingly, seeking catalysts that possessed low cost and high reaction activity is requisite for CO oxidation application.
Single-atom catalysis is considered an attractive strategy to reduce cost, make atom efficiency maximal, and then improve catalytic efficiency and selectivity, which has attracted much attention in environmental and energy catalysis [8-10]. Since Zhao [11] firstly reported the Pt single-atom catalyst that Pt1/FeOx possesses excellent stability and high activity for CO oxidation, remarkable advancements for single-atom catalysis have been made in various fields [12-15]. Numerous single-atom embellished catalysts have been fabricated and made great efforts in oxidation reaction [16-18], further confirming it can show desirable potentials in oxidizing poisonous CO [19, 20]. For example, Wang [21] prepared a single-atom Pd1/CeO2 catalyst by high-temperature atom trapping, and the catalyst shows excellent properties for low-temperature CO oxidation, and its active site was clarified. This work provides a new approach for tailoring the nature of active sites in metal single-atom catalysis by atom trapping.
An appropriate supporter is necessary for single-atom catalysis [22, 23]. Carbon-based materials have appeared much interest on account of their favorable physical and chemical performances [24, 25]. Among these, graphene including many advantages that excellent stability, conductivity, flexibility, and high specific surface areas [26-29], has been commonly acted as supporter material of catalysts [30-32] which can provide plenty of active sites.
The application of precious metals as single-atoms in single-atom catalytically oxidizing CO has developed rapidly, which lowers the content of noble metal as well the price of catalysts [33, 34]. There have been many reports about Pt or Au clusters for CO oxidation, some of which have benign catalytic performance [35, 36], so Pt and Au are selected as model single-atom in this article to study the role of single-atom catalysis in oxidizing CO. Whereas, there exist no agreement from the previous literature on whether Pt or Au single-atom has greater activities for CO oxidation, what roles do they play in CO oxidation reaction, and the reaction mechanism for oxidizing CO on single-atom decoration is controversial.
In short, the understanding of the CO oxidation reaction on Pt and Au single-atom catalysis remains vague, and thus appropriate means should be accomplished to explore its reaction processes, figure out rate-determining steps, and comprehend the role of the active atom. Herein, taking graphene as a model supporter, constructing Pt single-atom and Au single-atom supported graphene catalysts. Density functional theory (DFT) calculations were carried out to investigate the CO oxidation property on Pt and Au single-atom loaded graphene materials, and the potential mechanism was determined. Besides, calculating the minimum energy pathways (MEPs) and electronic structures, further studying the relationship between CO oxidation property and reactive atom. This work enhances the understanding of single-atom catalysis and can broaden its application ranges.
A periodical supercell of graphene containing 72 C atoms was carried out as a model supporter with a rectangle boundary (12.78 × 14.76 Å), and perpendicularly added 20 Å vacuum region to the graphene plane to minimize the interaction between different layers, seen in Fig. S1a (Supporting information) [37, 38]. Based on model graphene material that pristine graphene (GP), Pt and Au single-atom was introduced, labeled as Pt/GP and Au/GP, respectively, whose optimized structures were exhibited in Figs. S1b and c (Supporting information).
All the first-principle DFT calculations were carried out by utilizing the VASP5.4 code [39, 40] and the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) exchange and correlation functional [41]. The Gaussian smearing width was set to be 0.2 eV. A plane-wave basis set together with cut-off energy of 400 eV within the framework of the projector-augmented wave (PAW) method was employed [42, 43]. The van der Waals (vdW) correction was described by the D2 method of Grimme with default parameters. A 3 × 3 × 1 Monkhorst Pack grid of the Brillouin zone was sampled. All atoms, except those on the boundary, were allowed to relax and converge to 0.03 eV Å−1 for all systems. Searching minimum energy pathways (MEPs) is using the nudged elastic band (NEB) method [44, 45], which varies from an initial state (IS) to its final state (FS), using to define transitional state (TS) combined with climbing image method as well proved by a single imaginary frequency. The detailed reaction formulas of CO oxidation were listed in the Support Information.
The binding energy (ΔEb) is defined as (Eq. (1))
|
|
(1) |
where E(AmBn), m, E(A), n and E(B) are the total energy of the single-atom support graphene material, the numbers of C atom, the energy of single C atom, the numbers of single Pt or Au atom and the energy of single Pt or Au atom, respectively. As is known to all, the lower the binding energy, the more stable it is. The negative binding energy is exothermic.
Based on the binding energy formula, the results of the binding energy for Pt/GP and Au/GP were exhibited in Fig. S1. As shown in Fig. S1, the binding energy values for Pt/GP and Au/GP both are much more negative, hence Pt/GP and Au/GP materials both are stable and they give off heat during their formation.
The adsorption energy (ΔEads) is defined as (Eq. (2)):
|
|
(2) |
where Et, Es, and Em are the total energy of the adsorption complex, the graphene material, and the isolated molecule, respectively.
The single-atom decoration can affect the material property. Firstly, the charge distribution cases of the three graphene materials were investigated, and then the charge density difference and Bader analysis of the C atom, Pt atom, and Au atom were calculated. As shown in Fig. 1a, the C atom for pristine graphene (GP) was surrounded by blue areas with the Bader value of −0.12 e, which indicates it gains electrons. Whereas the Pt atom of Pt single-atom support graphene (Pt/GP) and the Au atom of Au single-atom support graphene (Au/GP) both were surrounded by yellow areas and the Bader values for they are similar and positive. Those indicate that the Pt and Au atoms lose electrons and present a positive center, which can attract negatively charged molecules.
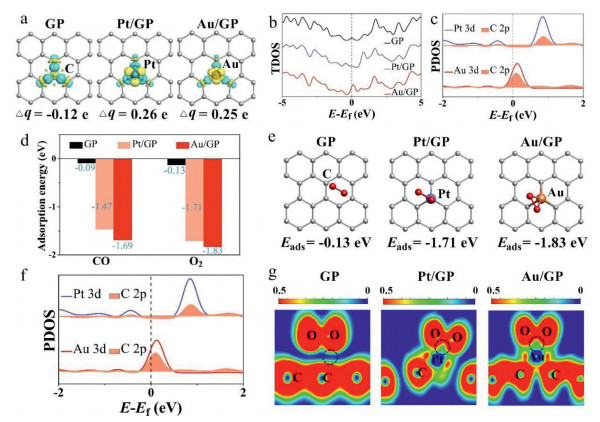
The total density of state (TDOS) for GP, Pt/GP, and Au/GP was implemented in Fig. 1b. Bringing in Pt and Au single-atom significantly influences the TDOS of graphene. In comparison with GP, the Pt atom of Pt/GP moved TDOS in the direction of the occupied state, indicating more electrons transfer on Pt/GP. As for Au/GP, the TDOS was optimized, and an active peak appeared around the Fermi level, which may assist in oxidizing CO. Accordingly, the introduction of a single-atom over graphene can improve electronic performances of the material and then enhance material catalytic properties. The interaction of the introduced single-atom and the surrounding C atom was further studied. As shown in Fig. 1c, there existed more overlapping areas at the Fermi level between Au 3d and C 2p than that between Pt 3d and C 2p, indicating a stronger mutual effect appeared on Au/GP, which implies the import of Au single-atom on graphene is more active than Pt.
Besides, the adsorption performances of CO and O2 on GP, Pt/GP, and Au/GP were investigated, possible adsorption constructions of CO and O2 on the three graphene materials (Fig. S2 for CO, Fig. S3 for O2 in Supporting information) were examined. The summarized histogram for adsorption energy of CO as well O2 on these materials was shown in Fig. 1d, and the values of adsorption energy were tabbed. Compared to GP, the adsorption energy of CO and O2 has immensely increased no matter on Pt/GP or Au/GP. O2 prefers to adsorb both on Pt/GP and Au/GP than CO, which is a vital step for subsequent CO oxidation. Thus, O2 adsorption performances over graphene materials were further discussed. It can be seen from Fig. 1e, O2 weekly adsorbed on GP with −0.13 eV energy, while Pt/GP and Au/GP attract O2 more with the adsorption values are −1.71 eV and −1.83 eV. The O2 adsorption site on GP is the center of the hexatomic ring, which for Pt/GP is bridged at the Pt atom and the center of the hexatomic ring, and that for Au/GP is atop the Au atom. Among which Au/GP possesses the largest adsorption energy of O2.
The partial density of state (PDOS) between the active atoms (the C, Pt, and Au atoms) and the O atom of adsorbed O2 was investigated (Fig. 1f). More overlapping zones existed between the Au 3d and the O 2p at around Fermi level (0 eV) and the range of −7 eV to −3 eV than that between the Pt and O as well the C and O, indicating the Au atom had the strongest interaction with the adsorbed O atom. The intensity of mutual effect between the adsorbed O and GP, Pt/GP as well Au/GP was further probed by electronic local function (ELF) analysis. It is seen in Fig. 1g, the adsorbed O possesses a much stronger interaction with Au/GP than GP and is not much different from that of Pt/GP. Consequently, the PDOS and ELF result concert with the adsorption results, which all reveal that Au/GP possesses the optimal O2 adsorption properties.
Eley-Rideal (ER) mechanism has two cases. 1) O2 adsorption state: O2 molecule directly participates in the reaction that is adsorbed and activated O2 reacts with free CO into CO2. 2) O2 dissociation state: surface activated O2 dissociates into oxygen atoms, and then the oxygen atoms severally react with free CO into CO2.
Based on the optimal O2 adsorption structures on the three graphene materials, the reaction processes of adsorbed O2 in molecular state directly reacting with CO to form CO2 in ER mechanism were determined through the minimum energy method. Relative potential energy and the side views of the optimized configurations of the initial state (IS), transition state (TS), final state (FS) at each step for the three graphene materials were denoted in Fig. 2.
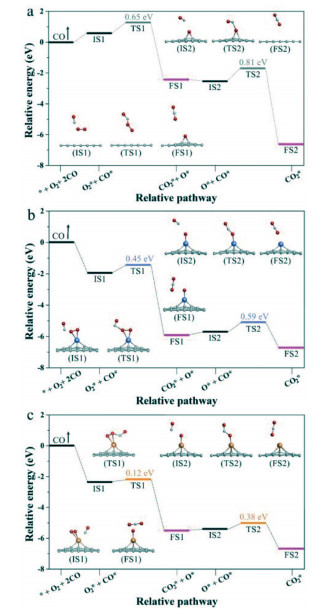
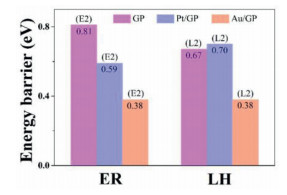
As shown in Fig. 2, adsorbed O2 attracts and initially oxide CO into CO2 + O* (tagged by 1H, and the following step is denoted as 2H). The 1H step proceeds on GP requires 0.65 eV energy barrier, while that on Pt/GP is 0.45 eV, and only 0.12 eV on Au/GP. The 2H step that the remained O* oxides CO to form CO2, which is the determining step for CO oxidation on the three graphene materials, where the activation energy of the 2H step is 0.81 eV for GP, 0.59 eV for Pt/GP, and 0.38 eV for Au/GP, respectively. In brief, CO oxidation processes in ER mechanism prefer to occur on Au/GP, which possesses the lowest energy barrier in oxidizing CO. Besides, the final product CO2 can desorb from Au/GP since it just calls for 0.14 eV desorption energy which can be overcome easily.
According to the MEPs of CO oxidation on the graphene materials, the 2H step is the reaction rate-limiting step in ER mechanism, and the activity order is Au/GP (0.38 eV) > Pt/GP (0.59 eV) > GP (0.81 eV), where Au/GP displays the most excellent property.
Based on the optimized adsorption structures of O2, O2 dissociation was researched, and reaction pathways of dissociation were shown in Fig. S4 (Supporting information). The activation barrier of O2 dissociation on GP is more than 3.00 eV, those of Pt/GP and Au/GP are close to 2.00 eV. It is observed that the lowest energy barrier of O2 dissociation on the graphene materials is desired to be 1.73 eV which is hard to accomplish, indicating that dissociating O2 on the graphene materials is kinetics not viable. Hence, the O2 dissociation ER mechanism for graphene materials is not considered in this article.
Furthermore, CO oxidation in Langmuir-Hinshelwood (LH) mechanism was carried out, and Fig. 3 shows the former step of the LH mechanism whose later step is the same as the 2H step of ER mechanism not be discussed repeatedly. Known from the picture, CO and O2 initial co-adsorption on the materials, then they react with each other to form OOCO* intermediate product, and further generate CO2 along with leaving an O* adsorbed on the material surface. The OOCO* adsorbs on the single-atom of Pt/GP and Au/GP and forms Pt/Au-O bond while OOCO* weak adsorption above GP and forms an epoxy group. As shown in Fig. 3, compared with GP, introducing Au single-atom decreases the value of the reaction rate-determining barrier by 43% but Pt increases a little. Accordingly, Au/GP can effectively improve CO oxidation performance in the LH mechanism which is far better than that of Pt/GP.
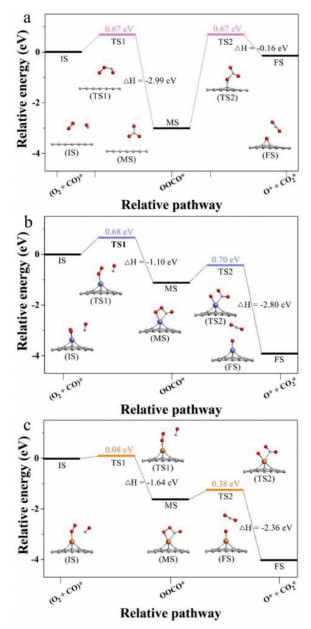
Based on the summary performance results of CO oxidation in the two mechanisms shown in Fig. 4, Au/GP shows the lowest activation barriers on the graphene materials, and the two mechanisms both are possible to follow. As for Pt/GP, it is observed to be the ER mechanism, as well GP is an LH mechanism.
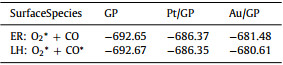
To further confirm the mechanism of the graphene materials to follow, the energy of adsorbed O2 with CO in ER mechanism (O2* + CO) and co-adsorbed O2 with CO in LH mechanism (O2* + CO*) on the three materials were made a comparison. As shown in Table 1, the energy value of O2* + CO on Au/GP is larger than O2* + CO*. The energy result of the two intermediate products for Au/GP indicates CO oxidation on Au/GP follows ER mechanism, which agrees with the activation energy result. However, the energy value of O2* + CO and O2* + CO* on GP is similar, same issues occur on Pt/GP, which indicate the energy results of the two intermediate products for GP and Pt/GP do not determine the choice of the reaction mechanism.
 DownLoad:
CSV
DownLoad:
CSV
|
Combining the oxidation performance with adsorption results, oxidizing CO on Au/GP and Pt/GP both follow ER mechanism but GP follows LH mechanism. Consequently, introducing a single-atom not only decreases the reaction barrier of graphene but also changes its reaction mechanism from LH mechanism into ER mechanism. Additionally, the introduction of Au single-atom is optimal.
As is well known, the conversion case from IS state to TS state, and the arrival of the transition state is early or late can reflect the speed of reaction rate. The excellent material Au/GP is inclined to follow ER mechanism and shows the best CO oxidation property. Hence, the transform cases from IS to TS at the rate-limiting step in ER mechanism of CO oxidation on the graphene materials were further analyzed. The detailed minimum energy pathways (MEPs) of CO oxidation in ER mechanism were demonstrated by Fig. S5 (Supporting information).
As mentioned above, the 2H step in ER mechanism is the rate-determining step of CO oxidation on Au/GP. However, the reason why Au/GP denotes the most satisfactory property for oxidizing CO than other graphene materials should be further discussed. It is seen in Fig. S5b, the length between the C atom of the absorbed CO (denoted as C1, the same below) and the O atom leftover from the absorbed O2 (denoted as O*, the same below) is 3.08 Å at the 2H step for GP, and shortens 1.28 Å to TS. Meanwhile, the distance between the O* and the C atom of GP (denoted as C2, the same below) extends from 1.46 Å for IS to 1.86 Å for TS, along with the variation of 0.40 Å. As for Pt/GP (Fig. S5d), the bond of the C1 and the O* prolongs 1.91 Å from IS to TS which requires 1.5 times change than that of GP, while the length between the O* and C2 just shortens 0.1 Å needing 1/4 times change compared to that of GP. As a result, Pt/GP can exhibit a better activity for the 2H step of CO oxidation than GP due to the early arrival of a transition state. Besides, Au demonstrates the best property for the 2H step of CO oxidation (Fig. S5f) owing to the earliest arrival of a transition state. The distance change between the C1 and the O* from IS to TS on Au/GP was 0.98 Å which is 4/5 times in contrast to GP, and that between the O* and C2 0.06 Å which is 3/20 times. As for the 2H step of CO oxidation in ER mechanism on the graphene materials, structural features meet CO oxidation performance results.
Furthermore, the electronic role of Au single-atom on graphene material should be investigated, enhancing the understanding of single-atom catalysis and broadening its application ranges. To our knowledge, the transition state is of importance for the reaction process, and its activation degree can reflect reaction activation energy measure and determine the reaction rate. Therefore, the TS of the 2H step for CO oxidation on GP and Au/GP were further discussed.
The charge density difference and Bader analysis for the adsorbed CO* and the adsorbed O* of the TS in the 2H step was investigated. As shown in Fig. 5a and b, the CO* and the O* (together denoted as COO*) on Au/GP was surrounded by more yellow and blue regions compared to that on GP, indicating the transferred electron numbers of the COO* on Au/GP are more than that on GP. Consistent with the charge density difference results, Bader analysis shows that the COO* on Au/GP owns more electrons than that on GP, accompanied by the Bader value is −0.66 e for Au/GP while that is −0.57 e for GP.
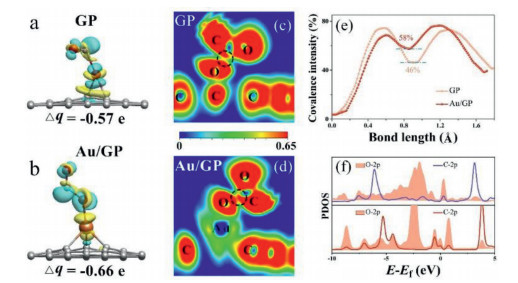
The ELF was explored to study the interaction between the adsorbed CO* and the adsorbed O* both on Au/GP and GP. As shown in Fig. 5c and d, there present yellow between the C atom of the CO* and the adsorbed O* on GP, while that is red on Au/GP, indicating the mutual effect between the C and the O atoms over Au/GP is stronger than that over GP, as well a stronger activation of the transition state. Besides, the line profiles of ELF for the TS in the 2H step on Au/GP and GP were calculated in Fig. 5e. It is found that the value of ELF between the C and the O is 58% on Au/GP and 46% on GP, indicating the atomic interaction on Au/GP is higher than that on GP. Accordingly, the adsorbed COO* is more intensely activated by Au/GP than GP.
Furthermore, the PDOS for the adsorbed CO* and the adsorbed O* over Au/GP and GP was calculated. As can be seen in Fig. 5f, there exist more overlapped areas between the C 2p orbital of the CO* and the O 2p orbital of the adsorbed O on Au/GP contrast with that on GP. The PDOS result shows that the adsorbed CO* and the adsorbed O* on Au/GP have a stronger interaction than that on GP, confirming Au/GP can activate TS strongly. This indicates Au/GP shows more excellent performances of CO oxidation. The result of electronic structure is consistent with the geometric structure, which both verify Au/GP is the optimum catalyst for CO oxidation over the graphene materials.
In conclusion, a first-principles calculation was utilized to investigate the reaction processes, rate-limiting steps, and reaction mechanism of single-atom support graphene in CO oxidation. Introducing Au single-atom into graphene can be an excellent strategy for improving CO oxidation property and changing its reaction mechanism from LH to ER mechanism, whose rate-determining barrier of CO oxidation is 0.38 eV, lower than 0.59 eV for Pt/GP and 0.81 eV for GP. The single-atom Au is the active center for adsorbing O2 and then oxidizing CO on Au/GP. The excellent properties in oxidizing CO on Au/GP are ascribed to the tailored density of state structures of the material, enhanced O2 adsorption performances, and improved activation of the reaction intermediate products. This work can afford an attractive understanding of single-atom catalysis in catalyzing CO and provide a guide on designing targeted catalysts with high reaction activity.
There are no conflicts to declare.
This work was supported by the National Key Research and Development Program of China (No. 2019YFC0214404), Science and Technology Major Projects in Sichuan Province (No. 2019KJT0067-2018SZDZX0019), and Science and Technology Major Projects in Chengdu (No. 2018-ZM01-00044-SN). The authors also acknowledge AM-HPC in Suzhou, China for computational support.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.032.
S. Sun, S. Wang, Y. Ye, B. Pan, Water Res. 153 (2019) 21–28.
doi: 10.1016/j.watres.2019.01.007
E. Rott, R. Minke, U. Bali, H. Steinmetz, Water Res. 122 (2017) 345–354.
doi: 10.1016/j.watres.2017.06.009
S. Wang, S. Sun, C. Shan, B. Pan, Water Res. 161 (2019) 78–88.
doi: 10.1016/j.watres.2019.05.099
X. Zhang, J. Li, W.Y. Fan, G.P. Sheng, Environ. Sci. Technol. 53 (2019) 4997–5004.
doi: 10.1021/acs.est.9b00348
Z. Wang, G. Chen, S. Patton, et al., Water Res. 159 (2019) 30–37.
doi: 10.1016/j.watres.2019.04.051
Q. Wang, Y. Shao, N. Gao, et al., Chem. Eng. J. 304 (2016) 201–208.
doi: 10.1016/j.cej.2016.06.092
R. Altaf, X. Lin, M.A. Tadda, S. Zhu, D. Liu, J. Clean. Prod. 278 (2021) 123960.
doi: 10.1016/j.jclepro.2020.123960
E. Rott, M. Nouri, C. Meyer, et al., Water Res. 145 (2018) 608–617.
doi: 10.1016/j.watres.2018.08.067
E. Rott, R. Minke, H. Steinmetz, J. Water Process Eng. 17 (2017) 188–196.
doi: 10.1016/j.jwpe.2017.04.008
Y. Liu, X. He, Y. Fu, D.D. Dionysiou, J. Hazard. Mater. 305 (2016) 229–239.
doi: 10.1016/j.jhazmat.2015.11.043
S. Wacławek, H.V. Lutze, K. Grübel, et al., Chem. Eng. J. 330 (2017) 44–62.
doi: 10.1016/j.cej.2017.07.132
Y.H. Guan, J. Ma, X.C. Li, J.Y. Fang, L.W. Chen, Environ. Sci. Technol. 45 (2011) 9308–9314.
doi: 10.1021/es2017363
X. Zhang, J. Yao, Z. Zhao, J. Liu, Chem. Eng. J. 364 (2019) 1–10.
doi: 10.1016/j.cej.2019.01.029
P. Hu, M. Long, Appl. Catal. B: Environ. 181 (2016) 103–117.
doi: 10.1016/j.apcatb.2015.07.024
B. Pan, Z. Li, Y. Zhang, et al., Chem. Eng. J. 248 (2014) 290–296.
doi: 10.1016/j.cej.2014.02.093
Y. Su, H. Cui, Q. Li, S. Gao, J.K. Shang, Water Res. 47 (2013) 5018–5026.
doi: 10.1016/j.watres.2013.05.044
L. Fang, B. Wu, I.M.C. Lo, Chem. Eng. J. 319 (2017) 258–267.
doi: 10.1016/j.cej.2017.03.012
D.P. Van Vuuren, A.F. Bouwman, A.H.W. Beusen, Glob. Environ. Chang. 20 (2010) 428–439.
doi: 10.1016/j.gloenvcha.2010.04.004
T. Zeng, X. Zhang, S. Wang, H. Niu, Y. Cai, Environ. Sci. Technol. 49 (2015) 2350–2357.
doi: 10.1021/es505014z
X. Liu, F. Huang, Y. Yu, et al., Appl. Catal. B: Environ. 253 (2019) 149–159.
doi: 10.1016/j.apcatb.2019.04.047
G.X. Huang, C.Y. Wang, C.W. Yang, P.C. Guo, H.Q. Yu, Environ. Sci. Technol. 51 (2017) 12611–12618.
doi: 10.1021/acs.est.7b03007
Q. Yi, J. Ji, B. Shen, et al., Environ. Sci. Technol. 53 (2019) 9725–9733.
doi: 10.1021/acs.est.9b01676
Y. Gao, Y. Zhu, L. Lyu, et al., Environ. Sci. Technol. 52 (2018) 14371–14380.
doi: 10.1021/acs.est.8b05246
J. Liu, J. Zhou, Z. Ding, et al., Ultrason. Sonochem. 34 (2017) 953–959.
doi: 10.1016/j.ultsonch.2016.08.005
C. Liu, L. Liu, X. Tian, et al., Appl. Catal. B: Environ. 255 (2019) 117763.
doi: 10.1016/j.apcatb.2019.117763
W.D. Oh, Z. Dong, T.T. Lim, Appl. Catal. B: Environ. 194 (2016) 169–201.
doi: 10.1016/j.apcatb.2016.04.003
A. Sarkar, S.K. Biswas, P. Pramanik, J. Mater. Chem. 20 (2010) 4417–4424.
doi: 10.1039/b925379c
S. Khan, X. He, J.A. Khan, et al., Chem. Eng. J. 318 (2017) 135–142.
doi: 10.1016/j.cej.2016.05.150
M. Li, B. Zhang, S. Zou, Q. Liu, M. Yang, J. Hazard. Mater. 384 (2020) 121386.
doi: 10.1016/j.jhazmat.2019.121386
B. Nowack, A.T. Stone, Water Res. 40 (2006) 2201–2209.
doi: 10.1016/j.watres.2006.03.018
S. Xiao, M. Cheng, H. Zhong, et al., Chem. Eng. J. 384 (2020) 123265.
doi: 10.1016/j.cej.2019.123265
Y. Wu, Y. Shi, H. Chen, J. Zhao, W. Dong, Process Saf. Environ. Prot. 116 (2018) 468–476.
doi: 10.1016/j.psep.2018.03.020
H. Gao, J. Chen, Y. Zhang, X. Zhou, Chem. Eng. J. 306 (2016) 522–530.
doi: 10.1016/j.cej.2016.07.080
S. Yang, P. Wang, X. Yang, et al., J. Hazard. Mater. 179 (2010) 552–558.
doi: 10.1016/j.jhazmat.2010.03.039
N. Liu, F. Ding, C.H. Weng, C.C. Hwang, Y.T. Lin, J. Environ. Manag. 206 (2018) 565–576.
doi: 10.1016/j.jenvman.2017.11.006
C. Luo, J. Ma, J. Jiang, et al., Water Res. 80 (2015) 99–108.
doi: 10.1016/j.watres.2015.05.019
L. Wang, W. Liu, T. Wang, J. Ni, Chem. Eng. J. 225 (2013) 153–163.
doi: 10.1016/j.cej.2013.03.081
R. Li, J.J. Wang, B. Zhou, et al., Sci. Total Environ. 559 (2016) 121–129.
doi: 10.1016/j.scitotenv.2016.03.151
H. Li, J. Wan, Y. Ma, Y. Wang, Z. Guan, RSC Adv. 5 (2015) 99935–99943.
doi: 10.1039/C5RA16094D
B. Lin, M. Hua, Y.Y. Zhang, et al., Chemosphere 166 (2017) 422–430.
doi: 10.1016/j.chemosphere.2016.09.104
J. Luo, X. Luo, J. Crittenden, et al., Environ. Sci. Technol. 49 (2015) 11115–11124.
doi: 10.1021/acs.est.5b02903
M. Mallet, K. Barthelemy, C. Ruby, A. Renard, S. Naille, J. Colloid Interface Sci. 407 (2013) 95–101.
doi: 10.1016/j.jcis.2013.06.049
S. Sun, S. Wang, Y. Ye, B. Pan, Water Res. 153 (2019) 21–28.
doi: 10.1016/j.watres.2019.01.007
E. Rott, R. Minke, U. Bali, H. Steinmetz, Water Res. 122 (2017) 345–354.
doi: 10.1016/j.watres.2017.06.009
S. Wang, S. Sun, C. Shan, B. Pan, Water Res. 161 (2019) 78–88.
doi: 10.1016/j.watres.2019.05.099
X. Zhang, J. Li, W.Y. Fan, G.P. Sheng, Environ. Sci. Technol. 53 (2019) 4997–5004.
doi: 10.1021/acs.est.9b00348
Z. Wang, G. Chen, S. Patton, et al., Water Res. 159 (2019) 30–37.
doi: 10.1016/j.watres.2019.04.051
Q. Wang, Y. Shao, N. Gao, et al., Chem. Eng. J. 304 (2016) 201–208.
doi: 10.1016/j.cej.2016.06.092
R. Altaf, X. Lin, M.A. Tadda, S. Zhu, D. Liu, J. Clean. Prod. 278 (2021) 123960.
doi: 10.1016/j.jclepro.2020.123960
E. Rott, M. Nouri, C. Meyer, et al., Water Res. 145 (2018) 608–617.
doi: 10.1016/j.watres.2018.08.067
E. Rott, R. Minke, H. Steinmetz, J. Water Process Eng. 17 (2017) 188–196.
doi: 10.1016/j.jwpe.2017.04.008
Y. Liu, X. He, Y. Fu, D.D. Dionysiou, J. Hazard. Mater. 305 (2016) 229–239.
doi: 10.1016/j.jhazmat.2015.11.043
S. Wacławek, H.V. Lutze, K. Grübel, et al., Chem. Eng. J. 330 (2017) 44–62.
doi: 10.1016/j.cej.2017.07.132
Y.H. Guan, J. Ma, X.C. Li, J.Y. Fang, L.W. Chen, Environ. Sci. Technol. 45 (2011) 9308–9314.
doi: 10.1021/es2017363
X. Zhang, J. Yao, Z. Zhao, J. Liu, Chem. Eng. J. 364 (2019) 1–10.
doi: 10.1016/j.cej.2019.01.029
P. Hu, M. Long, Appl. Catal. B: Environ. 181 (2016) 103–117.
doi: 10.1016/j.apcatb.2015.07.024
B. Pan, Z. Li, Y. Zhang, et al., Chem. Eng. J. 248 (2014) 290–296.
doi: 10.1016/j.cej.2014.02.093
Y. Su, H. Cui, Q. Li, S. Gao, J.K. Shang, Water Res. 47 (2013) 5018–5026.
doi: 10.1016/j.watres.2013.05.044
L. Fang, B. Wu, I.M.C. Lo, Chem. Eng. J. 319 (2017) 258–267.
doi: 10.1016/j.cej.2017.03.012
D.P. Van Vuuren, A.F. Bouwman, A.H.W. Beusen, Glob. Environ. Chang. 20 (2010) 428–439.
doi: 10.1016/j.gloenvcha.2010.04.004
T. Zeng, X. Zhang, S. Wang, H. Niu, Y. Cai, Environ. Sci. Technol. 49 (2015) 2350–2357.
doi: 10.1021/es505014z
X. Liu, F. Huang, Y. Yu, et al., Appl. Catal. B: Environ. 253 (2019) 149–159.
doi: 10.1016/j.apcatb.2019.04.047
G.X. Huang, C.Y. Wang, C.W. Yang, P.C. Guo, H.Q. Yu, Environ. Sci. Technol. 51 (2017) 12611–12618.
doi: 10.1021/acs.est.7b03007
Q. Yi, J. Ji, B. Shen, et al., Environ. Sci. Technol. 53 (2019) 9725–9733.
doi: 10.1021/acs.est.9b01676
Y. Gao, Y. Zhu, L. Lyu, et al., Environ. Sci. Technol. 52 (2018) 14371–14380.
doi: 10.1021/acs.est.8b05246
J. Liu, J. Zhou, Z. Ding, et al., Ultrason. Sonochem. 34 (2017) 953–959.
doi: 10.1016/j.ultsonch.2016.08.005
C. Liu, L. Liu, X. Tian, et al., Appl. Catal. B: Environ. 255 (2019) 117763.
doi: 10.1016/j.apcatb.2019.117763
W.D. Oh, Z. Dong, T.T. Lim, Appl. Catal. B: Environ. 194 (2016) 169–201.
doi: 10.1016/j.apcatb.2016.04.003
A. Sarkar, S.K. Biswas, P. Pramanik, J. Mater. Chem. 20 (2010) 4417–4424.
doi: 10.1039/b925379c
S. Khan, X. He, J.A. Khan, et al., Chem. Eng. J. 318 (2017) 135–142.
doi: 10.1016/j.cej.2016.05.150
M. Li, B. Zhang, S. Zou, Q. Liu, M. Yang, J. Hazard. Mater. 384 (2020) 121386.
doi: 10.1016/j.jhazmat.2019.121386
B. Nowack, A.T. Stone, Water Res. 40 (2006) 2201–2209.
doi: 10.1016/j.watres.2006.03.018
S. Xiao, M. Cheng, H. Zhong, et al., Chem. Eng. J. 384 (2020) 123265.
doi: 10.1016/j.cej.2019.123265
Y. Wu, Y. Shi, H. Chen, J. Zhao, W. Dong, Process Saf. Environ. Prot. 116 (2018) 468–476.
doi: 10.1016/j.psep.2018.03.020
H. Gao, J. Chen, Y. Zhang, X. Zhou, Chem. Eng. J. 306 (2016) 522–530.
doi: 10.1016/j.cej.2016.07.080
S. Yang, P. Wang, X. Yang, et al., J. Hazard. Mater. 179 (2010) 552–558.
doi: 10.1016/j.jhazmat.2010.03.039
N. Liu, F. Ding, C.H. Weng, C.C. Hwang, Y.T. Lin, J. Environ. Manag. 206 (2018) 565–576.
doi: 10.1016/j.jenvman.2017.11.006
C. Luo, J. Ma, J. Jiang, et al., Water Res. 80 (2015) 99–108.
doi: 10.1016/j.watres.2015.05.019
L. Wang, W. Liu, T. Wang, J. Ni, Chem. Eng. J. 225 (2013) 153–163.
doi: 10.1016/j.cej.2013.03.081
R. Li, J.J. Wang, B. Zhou, et al., Sci. Total Environ. 559 (2016) 121–129.
doi: 10.1016/j.scitotenv.2016.03.151
H. Li, J. Wan, Y. Ma, Y. Wang, Z. Guan, RSC Adv. 5 (2015) 99935–99943.
doi: 10.1039/C5RA16094D
B. Lin, M. Hua, Y.Y. Zhang, et al., Chemosphere 166 (2017) 422–430.
doi: 10.1016/j.chemosphere.2016.09.104
J. Luo, X. Luo, J. Crittenden, et al., Environ. Sci. Technol. 49 (2015) 11115–11124.
doi: 10.1021/acs.est.5b02903
M. Mallet, K. Barthelemy, C. Ruby, A. Renard, S. Naille, J. Colloid Interface Sci. 407 (2013) 95–101.
doi: 10.1016/j.jcis.2013.06.049

Huazhe Wang , Chenghuan Qiao , Chuchu Chen , Bing Liu , Juanshan Du , Qinglian Wu , Xiaochi Feng , Shuyan Zhan , Wan-Qian Guo . Synergistic adsorption and singlet oxygenation of humic acid on alkali-activated biochar via peroxymonosulfate activation. Chinese Chemical Letters, 2025, 36(5): 110244-. doi: 10.1016/j.cclet.2024.110244
Yu Yao , Jinqiang Zhang , Yantao Wang , Kunsheng Hu , Yangyang Yang , Zhongshuai Zhu , Shuang Zhong , Huayang Zhang , Shaobin Wang , Xiaoguang Duan . Nitrogen-rich carbon for catalytic activation of peroxymonosulfate towards green synthesis. Chinese Chemical Letters, 2024, 35(11): 109633-. doi: 10.1016/j.cclet.2024.109633
Yinyin Xu , Yuanyuan Li , Jingbo Feng , Chen Wang , Yan Zhang , Yukun Wang , Xiuwen Cheng . Covalent organic frameworks doped with manganese-metal organic framework for peroxymonosulfate activation. Chinese Chemical Letters, 2024, 35(4): 108838-. doi: 10.1016/j.cclet.2023.108838
Mengmeng Ao , Jian Wei , Chuan-Shu He , Heng Zhang , Zhaokun Xiong , Yonghui Song , Bo Lai . Insight into the activation of peroxymonosulfate by N-doped copper-based carbon for efficient degradation of organic pollutants: Synergy of nonradicals. Chinese Chemical Letters, 2025, 36(1): 109882-. doi: 10.1016/j.cclet.2024.109882
Yun-Xin Huang , Lin-Qian Yu , Ke-Yu Chen , Hao Wang , Shou-Yan Zhao , Bao-Cheng Huang , Ren-Cun Jin . Biochar with self-doped N to activate peroxymonosulfate for bisphenol-A degradation via electron transfer mechanism: The active edge graphitic N site. Chinese Chemical Letters, 2024, 35(9): 109437-. doi: 10.1016/j.cclet.2023.109437
Jiaxuan Wang , Tonghe Liu , Bingxiang Wang , Ziwei Li , Yuzhong Niu , Hou Chen , Ying Zhang . Synthesis of polyhydroxyl-capped PAMAM dendrimer/silica composites for the adsorption of aqueous Hg(II) and Ag(I). Chinese Chemical Letters, 2024, 35(12): 109900-. doi: 10.1016/j.cclet.2024.109900
Fengxing Liang , Yongzheng Zhu , Nannan Wang , Meiping Zhu , Huibing He , Yanqiu Zhu , Peikang Shen , Jinliang Zhu . Recent advances in copper-based materials for robust lithium polysulfides adsorption and catalytic conversion. Chinese Chemical Letters, 2024, 35(11): 109461-. doi: 10.1016/j.cclet.2023.109461
Congyan Liu , Xueyao Zhou , Fei Ye , Bin Jiang , Bo Liu . Confined electric field in nano-sized channels of ionic porous framework towards unique adsorption selectivity. Chinese Chemical Letters, 2025, 36(2): 109969-. doi: 10.1016/j.cclet.2024.109969
Ke Gong , Jinghan Liao , Jiangtao Lin , Quan Wang , Zhihua Wu , Liting Wang , Jiali Zhang , Yi Dong , Yourong Duan , Jianhua Chen . Mitochondria-targeted nanoparticles overcome chemoresistance via downregulating BACH1/CD47 axis in ovarian carcinoma. Chinese Chemical Letters, 2024, 35(5): 108888-. doi: 10.1016/j.cclet.2023.108888
Zixuan Zhu , Xianjin Shi , Yongfang Rao , Yu Huang . Recent progress of MgO-based materials in CO2 adsorption and conversion: Modification methods, reaction condition, and CO2 hydrogenation. Chinese Chemical Letters, 2024, 35(5): 108954-. doi: 10.1016/j.cclet.2023.108954
Yue Li , Minghao Fan , Conghui Wang , Yanxun Li , Xiang Yu , Jun Ding , Lei Yan , Lele Qiu , Yongcai Zhang , Longlu Wang . 3D layer-by-layer amorphous MoSx assembled from [Mo3S13]2- clusters for efficient removal of tetracycline: Synergy of adsorption and photo-assisted PMS activation. Chinese Chemical Letters, 2024, 35(9): 109764-. doi: 10.1016/j.cclet.2024.109764
Maosen Xu , Pengfei Zhu , Qinghong Cai , Meichun Bu , Chenghua Zhang , Hong Wu , Youzhou He , Min Fu , Siqi Li , Xingyan Liu . In-situ fabrication of TiO2/NH2−MIL-125(Ti) via MOF-driven strategy to promote efficient interfacial effects for enhancing photocatalytic NO removal activity. Chinese Chemical Letters, 2024, 35(10): 109524-. doi: 10.1016/j.cclet.2024.109524
Mengfan Zhang , Lingyan Liu , Peng Wei , Wei Feng , Tao Yi . A proximity tagging strategy utilizing an activated aldehyde group as the active site. Chinese Chemical Letters, 2025, 36(4): 110127-. doi: 10.1016/j.cclet.2024.110127
Yusong Bi , Rongzhen Zhang , Kaikai Niu , Shengsheng Yu , Hui Liu , Lingbao Xing . Construction of a three-step sequential energy transfer system with selective enhancement of superoxide anion radicals for photocatalysis. Chinese Chemical Letters, 2025, 36(5): 110311-. doi: 10.1016/j.cclet.2024.110311
Chong Liu , Nanthi Bolan , Anushka Upamali Rajapaksha , Hailong Wang , Paramasivan Balasubramanian , Pengyan Zhang , Xuan Cuong Nguyen , Fayong Li . Critical review of biochar for the removal of emerging inorganic pollutants from wastewater. Chinese Chemical Letters, 2025, 36(2): 109960-. doi: 10.1016/j.cclet.2024.109960
Dong Cheng , Youyou Feng , Bingxi Feng , Ke Wang , Guoxin Song , Gen Wang , Xiaoli Cheng , Yonghui Deng , Jing Wei . Polyphenol-mediated interfacial deposition strategy for supported manganese oxide catalysts with excellent pollutant degradation performance. Chinese Chemical Letters, 2024, 35(5): 108623-. doi: 10.1016/j.cclet.2023.108623
Kexin Yin , Jingren Yang , Yanwei Li , Qian Li , Xing Xu . Metal-free diatomaceous carbon-based catalyst for ultrafast and anti-interference Fenton-like oxidation. Chinese Chemical Letters, 2024, 35(12): 109847-. doi: 10.1016/j.cclet.2024.109847
Ming-Zhen Li , Yang Zhang , Kun Li , Ya-Nan Shang , Yi-Zhen Zhang , Yu-Jiao Kan , Zhi-Yang Jiao , Yu-Yuan Han , Xiao-Qiang Cao . In situ regeneration of catalyst for Fenton-like degradation by photogenerated electron transportation: Characterization, performance and mechanism comparison. Chinese Chemical Letters, 2025, 36(1): 109885-. doi: 10.1016/j.cclet.2024.109885
Jiayi Guo , Liangxiong Ling , Qinwei Lu , Yi Zhou , Xubiao Luo , Yanbo Zhou . Degradation of chloroxylenol by CoSx activated peroxomonosulfate: Role of cobalt-sulfur ratio. Chinese Chemical Letters, 2025, 36(4): 110380-. doi: 10.1016/j.cclet.2024.110380
Qingbai Tian , BingLiang Yu , Zhihao Li , Wei Hong , Qian Li , Xing Xu . Versatile catalytic membranes anchored with metal-nitrogen based metal oxides for ultrafast Fenton-like oxidation. Chinese Chemical Letters, 2025, 36(6): 110322-. doi: 10.1016/j.cclet.2024.110322
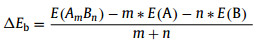
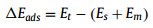
 DownLoad:
DownLoad: