The abnormal activation of JAK2 kinase is closely related to the occurrence and progression of myeloproliferative neoplasms (MPNs). At present, there is still an obvious unmet medical need for selective JAK2 inhibitors in clinic. In this paper, a class of 2-aminopyridine derivatives as potent and selective JAK2 inhibitors was obtained by combining drug design, synthesis and structure-activity relationship studies based on the previously identified lead Crizotinib. Among them, 21b exhibited high inhibitory activity against JAK2 with an IC50 of 9 nmol/L, moreover, it showed 276- and 184-fold selectivity over JAK1 and JAK3, respectively. Besides, 21b had a significant antiproliferative activity against HEL cells, and also inhibited the phosphorylation of JAK2 and its down-stream signaling pathway. These results indicated that 2-aminopyridine compound 21b had the potential to be developed as a selective JAK2 inhibitor for further study.
Janus kinases include JAK1, JAK2, JAK3 and TYK2, which belong to the family of nonreceptor tyrosine kinases. The Janus family regulates signal transduction through JAK-STAT (STAT, signal transductions and activators of transcription) signaling pathway, and plays an essential role in innate and adaptive immunity and hematopoiesis [1, 2]. A variety of cytokines activates Janus kinases, causing phosphorylation and dimerization of STAT proteins in the cell, and then transfer to the nucleus to regulate gene transcription [3, 4]. JAK2 mediates the signal transduction of cytokines such as IL-3, IL-5, granulocyte macrophage colony stimulating factor (GM-CSF), erythropoietin (EPO) and thrombopoietin (TPO), which are related to the growth and progression of myeloid cells [5-7]. Therefore, the excessive activity of JAK2 causes the JAK2-STAT signaling pathway to be constitutively activated, and leading to a variety of malignant diseases [8].
Myeloproliferative neoplasms (MPNs) include chronic myeloid leukemia (CML), myelofibrosis (MF), essential thrombocythemia (ET), and polycythemia vera (PV), which are a group of heterogeneous hematologic diseases resulting from deregulated proliferation of myeloid cells [9]. JAK2-STAT can be abnormally activated by mutations in JAK2 pseudokinase domain (JAK2 V617F), thrombopoietin receptor (MPL W515l) and calreticulin gene (CALR) exon 9, causing the occurrence and progression of myeloproliferation neoplasms (MPNs) [10, 11]. The V617F mutation in JAK2 was found to be carried by approximately 95% of PV patients and approximately 50% of MF and ET patients [12].
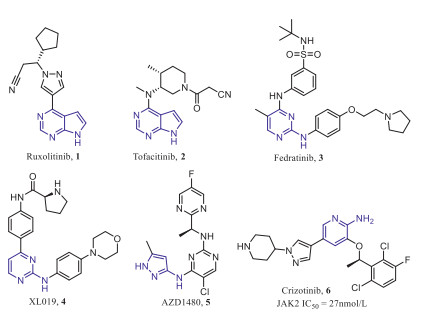
Indeed, several pan-JAK inhibitors have been marketed and selective JAK2 inhibitors continue to be discovered (Fig. 1). The pan-JAK inhibitors Ruxolitinib (1) and Tofacitinib (2) were approved for the treatment of myelofibrosis and rheumatoid arthritis, respectively [13-15]. Fedratinib (3) is currently the only approved selective JAK2 inhibitor for the treatment of primary or secondary (intermediate-2 or high-risk) myelofibrosis [16]. Unfortunately, there are several selective JAK2 inhibitors, such as XL019 (4) and AZD1480 (5), clinical trials of which were suspended due to central or peripheral neurotoxicity and dose-limiting toxicity, respectively [17-19]. These results indicate that the development of selective JAK2 inhibitors over the broader kinase spectrum and other JAK family kinases can reduce off-target side effects and unnecessary immunosuppression [20]. However, the high degree of homology in catalytic domain of the JAK family makes the discovery of selective inhibitors against JAK2 proved to be a huge challenge [21, 22].
Figure 1
Figure 1.
Structures of representative JAK2 inhibitors.
Our lab has been committed to discovering and developing JAK2 selective inhibitors as anticancer drugs, and found that ALK and c-MET inhibitor Crizotinib (Fig. 1) had good inhibitory activity against JAK2 with an IC50 value of 27 nmol/L, then, in order to improve the activity and selectivity of JAK2, the benzene ring in the hydrophobic region and the solvent exposed region of Crizotinib were optimized, and some effective compounds were obtained [23-25], however, these compounds have not been further studied due to druggability or toxicity problems.
In this paper, we focused on exploring a wider range of pharmacophore effects on JAK2 activity and selectivity through structure-based drug design. A series of 2-aminopyridine structures derived from Crizotinib were designed and synthesized as JAK2 selective inhibitors. The in vitro kinase inhibitory activities and selectivity of compounds were evaluated, and the cell antiproliferative activity and primary mechanism of the most potential compound were analyzed.
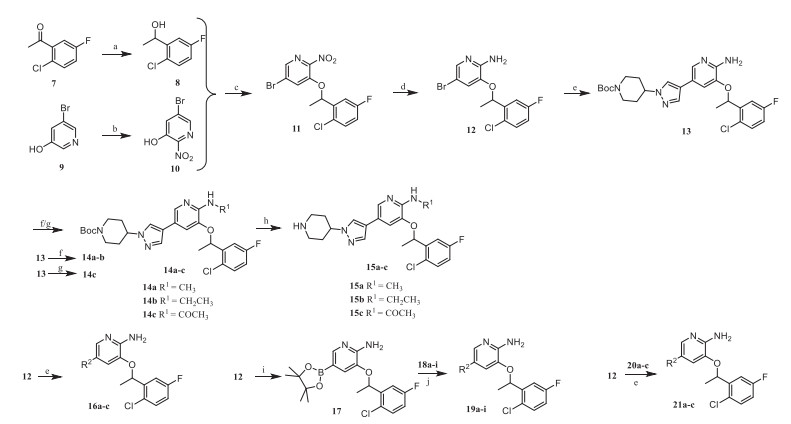
The general synthetic route of compounds 15a-c, 16a-c, 19a-i and 21a-c was shown in Scheme 1. The commercially available starting material 2‑chloro-5-fluoroacetophenone was reduced by NaBH4 to obtain 8, which was subjected to Mitsunobu reaction with intermediate 10 obtained by nitration of 3‑hydroxy-5-bromopyridine to obtain intermediate 11. The key intermediate 12 was obtained after 11 was reduced by Fe powder, then it was coupled with aryl borate to obtain intermediate 13, after which a substitution reaction occurred on 13. Compounds 14a-c were treated with trifluoroacetic acid to obtain the target products 15a-c. The target products 16a-c and 21a-c were obtained by Suzuki coupling of the key intermediate 12 with different aryl borates. Compound 12 could also be coupled with boric acid ester first, and then coupled with aryl halides to obtain the target products 19a-i.
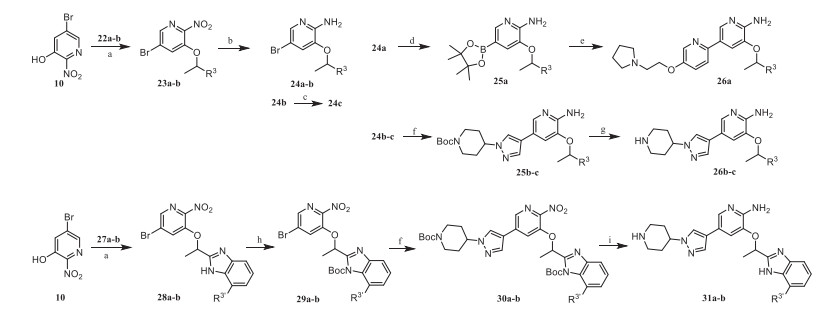
Compounds 26a-c, 31a and 31b were synthesized according to Scheme 2. Intermediates 23a and 23b were prepared by Mitsunobu reaction with 10 and 1-aryl-1-ethanol, which were reduced by Fe powder to obtain 24a and 24b, then cyano group of 24b was reduced by NaBH4 to obtain the amide derivative 24c. Intermediate 24a was coupled with boric acid ester and then coupled with aryl halide to obtain the target product 26a, while 24b and 24c were coupled with aryl borate and treated with trifluoroacetic acid to obtain the target products 26b and 26c. The intermediate 28a and 28b were obtained by the Mitsunobu reaction of 10 and benzimidazole derivatives. Compounds 28a and 28b were protected by Boc group and then coupled with aryl borate to obtain intermediates 30a and 30b. Finally, 31a and 31b were provided by reducing 30a and 30b and removing the Boc group.
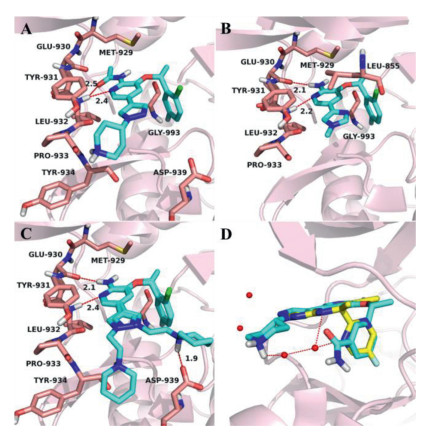
The pharmacophore of Crizotinib consist of a hydrophobic 2, 6-dichloro-3-fluorophenyl group, a 2-aminopyridine core located in the hinge region and a solvent exposed region. In order to explore SAR, we first introduced some small substituents on the amino group. However, from the enzymatic activity results (Table 1), the activities of compounds 15a and 15b were greatly reduced compared to Crizotinib. The results indicated that these substituents may collide with the gatekeeper residue Met929 in the ATP binding pocket of JAK2, thereby affecting the interaction with the hinge region. The potency of 15c was better than 15a and 15c, because the carbonyl oxygen atom on the acetyl group faced to the hinge region and probably formed hydrogen bond interaction with Leu932, but the key hydrogen bond interaction between the amino and the hinge region Glu930 disappeared (Fig. 2A).
Table 1
Table 1.
SAR exploration of different substituents in 2-aminopyridine.
Figure 2.
The docking poses of 15c (A), 19a (B), 21b (C) and 26c (D) in the JAK2 ATP-binding pocket (PDB: 2XA4). The receptor is shown in pink cartoon, 15c, 19a, 21b and 26c are presented as blue stick model. (A) The carbonyl oxygen atom and pyridine nitrogenatom of 15c formed H-bonds with the hinge residue Leu932. (B) The 2-aminopyridine scaffold of 19a can form two H-bonds with the hinge residues Glu930 and Leu932. (C) The 2-aminopyridine scaffold of 21b was firmly bound by two hydrogen bonds with the hinge residues Glu930 and Leu932, while 2-piperidinylethyl could swing to the left and right in the solvent region, forming an additional hydrogen bond with Asp939 when it pointed to the right. (D) Superposition of 26c and AZD1480 in JAK2 ATP-binding pocket, AZD1480 is shown in yellow stick model.
Subsequently, the efforts were paid to the modification of the solvent exposed region, in which the non-conserved residues are considered to be the main factors for improving selectivity [26]. In our previous work, we mainly explored the influence of six-membered aromatic rings and five-fused-six membered heterocyclic rings on selectivity [25], while in this paper, we focused on the effects of five-membered aromatic rings and longer hydrophilic side chains. In order to explore the influence of the position and number of methyl substitutions on pyrazole, we first designed compounds 16a-c. The results showed that the 16a with a methyl group at 1-position of pyrazole had the best inhibitory potency for JAK2 (IC50 = 0.024 µmol/L), while the activities of 16b and 16c decreased significantly. The possible reason was the introduction of methyl substitutions at both 3-position and 5-position, which destroyed the stability of the ligand planar arrangement. At the same time, compounds 19a and 19b were designed to explore the effect of the position and number of nitrogen atoms of five-membered aromatic heterocycles on the activity. Compared to 16a, when the pyrazole ring was replaced by the imidazole ring, the potency of 19a against JAK2 was 77-fold lost. The similar tendency was also found between compounds 16a and 19b. According to the docking pose of 19a (Fig. 2B), the carbonyl oxygen atoms of Leu932 and Leu855 faced to the ortho-position of the five-membered heterocycle, therefore, the nitrogen atoms at this position (19a, 19b) may had an unfavorable electrostatic interaction with carbonyl oxygen atoms and reduced the activity.
After the two rounds of optimization described above, compared with Crizotinib, their inhibitory activities against JAK2 were still not significantly improved due to the absence of additional beneficial interactions. Therefore, we hypothesized that the pyrazole of 16a was replaced with six-membered aromatic ring containing lactam or cyano substituents so that it could interact with the hinge region Tyr931 by hydrogen bonding. Immediately, compounds 19c-e were designed and synthesized, but their potencies were reduced remarkably compared with 16a. This result implied that the six-membered aromatic ring plane failed to rotate towards Tyr931, thus the carbonyl or cyano group could not form hydrogen bonds with the hinge region as we expected.
The 19f obtained by replacing pyrazole with pyridine has better activity, which stimulated us to investigate the active potential of pyridine modification. In addition, the substitution modification of pyridine is easier to synthesize than pyrazole, so the influence of hydrophilic side chain of pyridine on the activity was explored next. After comparing among 19g, 19h and 19i, it was concluded that the 3-position (2-pyrrolidine)ethoxy of pyridine was superior to 4-position, and the activity was better when the nitrogen atom of pyridine was adjacent to the scaffold. In general, the activities of compounds 19g-i were still significantly lower than that of 19f. The possible reason was the secondary amine group on the piperazine of 19f could interact with water molecules in the solvent region, but the side chains of 19g-i were too large to bind with the target. Hence, we had to reinvestigate the effect of the side chains with moderate length on activity in the presence of the pyrazole group. Surprisingly, compounds 21a-c showed very good activity, especially 21b, with an IC50 value of 0.009 ± 0.001 µmol/L. In the binding pocket of JAK2 (Fig. 2C), the 2-aminopyridine scaffold of 21b was firmly bound by two hydrogen bonds with the hinge residues Glu930 and Leu932; the halogen-substituted benzene occupied the hydrophobic cavity; 2-piperidinylethyl could swing to the left and right in the solvent region, forming an additional hydrogen bond with Asp939 when it pointed to the right, which may be the main reason for its increased activity.
Finally, we turned our attention to the benzene ring in the hydrophobic region to study its effect on activity. We designed compound 26a without the F and Cl substituents on the benzene, in order to change the overall trend of the molecule to make it closer to the hinge region. However, the potency of 26a was reduced by 24-fold compared to 19g, indicating that the halogens might play an important role in the binding of the benzene and the hydrophobic region. We analyzed the superposition of JAK2 inhibitor AZD1480 and 26c in the JAK2 pocket (Fig. 2D), and found that adding a cyano group and an amide group to the 3-position of the benzene as hydrogen bond donors might interact with the water molecule. On the contrary, the activity of 26b and 26c did not improve as estimated, it is speculated that the expected hydrogen bond interaction did not occur, and the presence of large groups would also cause desolvation and reduced the activity. We also substituted benzene with benzimidazole and designed compounds 31a and 31b, but their activity was completely lost. Although the hydrophobic pocket could accommodate benzimidazole, it formed a torsion angle compared with the benzene (Fig. S1 in Supporting information), which might be the main cause of inactivation.
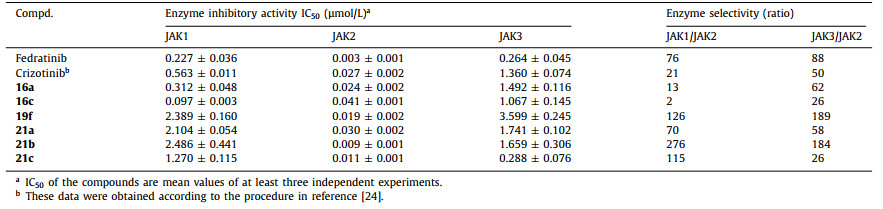
Subsequently, several compounds (16a, 16c, 19f, 21a, 21b and 21c) with better JAK2 inhibitory activity were selected for selective testing of JAK1 and JAK3. As indicated in Table 2, these compounds exhibited high JAK2 selectivity over JAK1 and JAK3, among which 21b was 276- and 184-fold selective over JAK1 and JAK3, respectively. This series of compounds exhibited weak inhibitory activity against JAK3 probably due to the influence of Ala966 in the hydrophobic pocket area, while there is a Gly993 in JAK2. In addition, the piperidine nitrogen atom of 21b formed a hydrogen bond with the solvent area Asp939 of JAK2, instead of Glu966 in JAK1, which explained its good selectivity. The results also indicated that substituents in the solvent region could significantly affect the selectivity and inhibitory activity of JAK enzyme.
Table 2
Table 2.
Selective test of compounds against JAK1, JAK2 and JAK3.
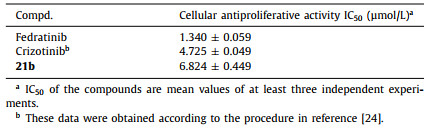
Based on the results of enzyme activity assays, 21b was further evaluated for its antiproliferative activity against the HEL cells (Table 3), which is known as human erythroleukemia cell line contains JAK2 V617F mutation. The results showed that compound 21b could significantly inhibit the proliferation of HEL cells with an IC50 value of 6.824 µmol/L.
Table 3
Table 3.
The antiproliferative activity on HEL cells of 21b.
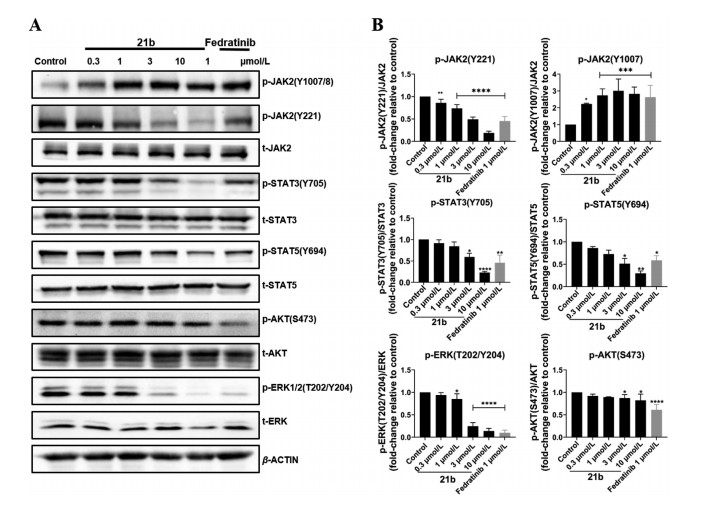
In order to explore the effect of 21b in the JAK2-STAT signal transduction pathways in HEL cells, Western-blot experiments were performed to assay its effect on the phosphorylation level of JAK2 and its downstream targets. As shown in Fig. 3, compound 21b dose-dependently increased the phosphorylation of JAK2(Y1007/8), which is consistent with the reported type I JAK2 inhibitors. Moreover, 21b inhibited JAK2(Y221), STAT3(Y705), STAT5(Y694) and ERK(T202/Y204) phosphorylation in a dose-dependent manner. In addition, 21b showed weakly inhibition on AKT phosphorylation. In short, these results indicated that 21b inhibited the activation of the JAK2-STAT signaling pathway by blocking the phosphorylation of JAK2 and its downstream substrates in HEL cells.
Figure 3
Figure 3.
Compound 21b effectively blocked the JAK2-STAT signaling pathways in HEL cells. (A) Western blot analysis of phosphorylation of JAK2 (Y1007/8), JAK2 (Y221), STAT3, STAT5, AKT and ERK. (B) The bands intensity was quantitatively analyzed from Western blotting images. Each column represents the means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In summary, based on the structure of Crizotinib, some potential selective JAK2 inhibitors were designed by optimizing the 2-aminopyridine scaffold, the kinase solvent region and the hydrophobic region, respectively. After structure confirmation and systematic biological activity evaluation, 21b showed high inhibitory activity against JAK2 with an IC50 of 9 nmol/L, moreover, it showed 276- and 184-fold selectivity over JAK1 and JAK3, respectively. 21b significantly inhibited the proliferation of HEL cells with an IC50 of 6.824 µmol/L, and blocked JAK2-STAT signaling pathway in HEL cells. The results indicated that 2-aminopyridine compound 21b was expected to be developed as a selective JAK2 inhibitor, and the structure-activity analysis process will be beneficial to the development of new selective JAK2 inhibitors.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The research is supported in part by the National Key Research and Development Program (No. 2016YFA0502304), the National Natural Science Foundation of China (No. 81825020), the Shanghai Committee of Science and Technology (No. 20S11901000), the Fundamental Research Funds for the Central Universities. Honglin Li is also sponsored by the National Program for Special Supports of Eminent Professionals and the National Program for Support of Top-Notch Young Professionals.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.099.
Qi Wang
, Yicong Gao
, Feng Lu
, Quli Fan
. Preparation and Performance Characterization of the Second Near-Infrared Phototheranostic Probe: A New Design and Teaching Practice of Polymer Chemistry Comprehensive Experiment. University Chemistry,
2024, 39(11): 342-349.
doi: 10.12461/PKU.DXHX202404141
Figure 2. The docking poses of 15c (A), 19a (B), 21b (C) and 26c (D) in the JAK2 ATP-binding pocket (PDB: 2XA4). The receptor is shown in pink cartoon, 15c, 19a, 21b and 26c are presented as blue stick model. (A) The carbonyl oxygen atom and pyridine nitrogenatom of 15c formed H-bonds with the hinge residue Leu932. (B) The 2-aminopyridine scaffold of 19a can form two H-bonds with the hinge residues Glu930 and Leu932. (C) The 2-aminopyridine scaffold of 21b was firmly bound by two hydrogen bonds with the hinge residues Glu930 and Leu932, while 2-piperidinylethyl could swing to the left and right in the solvent region, forming an additional hydrogen bond with Asp939 when it pointed to the right. (D) Superposition of 26c and AZD1480 in JAK2 ATP-binding pocket, AZD1480 is shown in yellow stick model.
Figure 3. Compound 21b effectively blocked the JAK2-STAT signaling pathways in HEL cells. (A) Western blot analysis of phosphorylation of JAK2 (Y1007/8), JAK2 (Y221), STAT3, STAT5, AKT and ERK. (B) The bands intensity was quantitatively analyzed from Western blotting images. Each column represents the means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.

 Login In
Login In




 DownLoad:
DownLoad:


 DownLoad:
DownLoad: