
 Login In
Login InInsight into the photoexcitation effect on the catalytic activation of H2 and C-H bonds on TiO2(110) surface
-
* Corresponding author.
E-mail address: hfwang@ecust.edu.cn (H.-F. Wang).
Figures(4)
Citation:
Min Zhou, Hai-Feng Wang. Insight into the photoexcitation effect on the catalytic activation of H2 and C-H bonds on TiO2(110) surface[J]. Chinese Chemical Letters,
;2022, 33(10): 4705-4709.
doi:
10.1016/j.cclet.2021.12.074

Figures(4)




The activation of H–H and C–H bonds (classified as R-H, R = H, C) is of vital importance in the hydrogenation and dehydrogenation processes for the catalytic conversion and functionalization of saturated or unsaturated hydrocarbon compounds. Ways to efficiently break these chemical bonds under mild condition are always at the heart of chemical industry [1-3], which constitutes one of the foundations for the controllable formation of value-added commodity [4-6]. Generally, the catalytic cleavage and functionalization of R-H bond have been extensively explored on kinds of catalysts, such as the noble metals (Pt [7-9], Rh [10-12], Au [13, 14] and et al. [15-18]), transition metal oxides (TMOs) and zeolites [19-21]. For example, the TMO-catalysts modified by single-atom metal doping or metal loading [22-26] were specifically manufactured to tailor the catalytic activity toward R-H bond activation. Despite these great progresses, these reactions are often performed at high temperatures (573–873 K) [27-29], which could lead to severe coke deposition and low product selectivity. Therefore, it is of paramount importance to develop more efficient catalysts or technologies for R-H bond activation under mild condition [30-33].
In this regard, the photocatalytic technology is believed to be capable of altering the reaction kinetics under mild condition owing to the presence of the excited carrier (h+/e−), and increasing experimental studies have demonstrated the feasibility for facilitating the R-H bond activation [34-36]. Taking the photocatalytic CH4 activation as an example, it was proposed that CH4 can be selectively converted to CH3OH or HCHO under UV irradiation at room temperature over semiconductor catalysts, like TiO2 [37], ZnO [38] and et al. [39-41]. However, the atomic-level insight into the photocatalytic effect is not unambiguously disclosed, to the best of our knowledge. Theoretically, owing to the difficulty in simulating photo-excited radicals properly on the semiconductor, the photocatalytic surface reaction has been relatively less studied with the detailed kinetic information provided [42-44]. It seems that most researches mainly focus on thermodynamic properties to evaluate the photocatalytic activity by considering the band structure of semiconductor and the potential of target reaction [45-48]. Kinetically, a comprehensive understanding of the nature of photocatalytic effect on R-H bond activation at the atomic-scale level is very limited. Specifically, one may ask, how does the photoexcitation affect the H2 and C-H activation, and what is the quantitative difference between thermo- and photo-catalytic mechanisms? Moreover, there are different types of C-H bonds, e.g., C(sp3)-H, C(sp2)-H and C(sp1)-H, and what are the general rule or difference of the photocatalytic effects on them?
Herein, we computationally explored the activation of H2 molecule and diverse C–H bonds in thermo- and photo-catalysis on rutile TiO2(110), one of the most studied semiconductors in photocatalysis. The theoretical results explicitly revealed the reaction mechanisms of H2 activation; moreover, the activation of C-H bonds in diverse light-hydrocarbons, including acetylene (HC≡C–H), benzene (C6H5–H), ethylene (CH2=CH–H), methane (CH3–H), ethane (CH3CH2–H), propyne (CH≡CCH2–H), toluene (C6H5CH2–H) and propylene (CH2=CHCH2–H), were calculated and compared. The C–H bond energy (Ebond) and deprotonation energy (Edep, corresponding to the acidity) were identified to be two main factors leading to the distinct behaviors of R–H bonds activation in thermo- and photo-excited conditions.
All spin-polarized DFT calculations were performed via Vienna ad initio Software Package (VASP) code [49, 50], using the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) [51, 52]. The project-augmented wave (PAW) [53] method was used to represent the core-valence electron interaction, and the valence electronic states were expanded in plane wave basis sets with a cutoff energy of 450 eV. To ensure proper description of the valence electrons on Ti atom, a valence electron configurations 3p64s23d2 (small core) was applied.
We adopted a p(3 × 2) model of rutile TiO2(110) with four TiO2 atomic layers to accommodate the organic molecule and minimize the interaction between periodically repeated slabs. The vacuum between slabs was ~10 Å and a 2 × 3 × 1 k-point mesh was used during the optimizations. The transition states (TSs) were searched by the constrained optimization scheme and were verified when (i) all forces on the atoms vanish and (ii) the total energy is a maximum along the reaction coordinate but a minimum with respect to the rest of the degrees of freedom [54-56]. All the atoms were allowed to relax, until the force on each atom was less than 0.05 eV/Å. The van der Waals forces was taken into account throughout this work to consider the possible weak interaction between intermediates and TiO2(110) surface [57, 58]. To model the photogenerated h+/e−, here we took the method of removing/adding electrons directly from the system with an additional neutralizing background charge applied in VASP due to the large dielectric constant of rutile TiO2 [59]. Moreover, the photogenerated carriers usually tend to delocalize on TiO2 system in GGA calculations due to the self-interaction error. To overcome this issue, DFT+U method with an on-site Coulomb interaction U added to the Ti-3d (U = 4.2 eV) and O-2p orbitals (U = 6.3 eV) was adopted for structure optimization and energy calculation [60], and the HSE06 hybrid functional was used to verify the electronic structure. Such an approach has been successfully applied to describe the photocatalytic reactions (e.g., methanol oxidation, oxygen evolution reaction) on TiO2 [61, 62].
The adsorption energy of surface species (X) was defined as Ead(X) = E(X/sur) – E(sur) – E(X), where E(sur), E(X) and E(X/sur) are the energy of catalyst surface, X species in the gas phase and X species adsorbed on the catalyst surface, respectively. Notably, at a specific temperature, the large entropy contributions of gaseous molecules were considered to estimate the adsorption free energy (Gad) [63, 64].
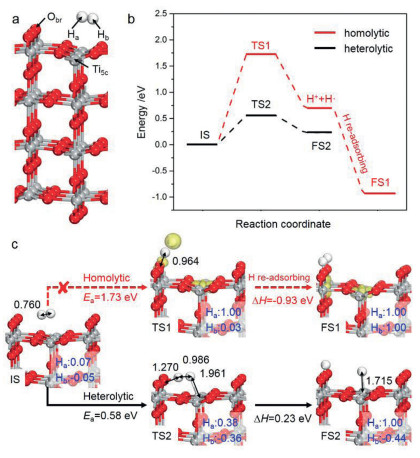
As Fig. 1a shows, the rutile TiO2(110) surface is terminated by two types of unsaturated sites: two-fold coordinated bridge O and five-fold coordinated Ti (denoted as Obr and Ti5c, respectively), which constitute the possible active centers for H-H and C-H bonds activation. Firstly, the adsorption and dissociation of H2 molecule under the thermocatalytic condition were briefly examined as a benchmark. It was found that H2 can weakly adsorb on Ti5c site with an adsorption energy, Ead(H2), of −0.20 eV, which corresponds to an adsorption free energy, Gad(H2), of 0.20 eV at room temperature. Subsequently, we tested two possible H2 dissociation routes, including homolytic and heterolytic cleavage pathways, according to previous studies [30, 31, 65]. In the homolytic mode, H2 activation is accompanied by the formation of a H· and a H+ (i.e., H2 + Ti5c4+ + Obr2− → H· + Ti5c3+ + ObrH−); the activation barrier is as high as 1.73 eV and the enthalpy change reaches 0.70 eV (see energy profiles in Fig. 1b). Alternatively, when the reaction proceeds via the heterolytic cleavage mode, which synergistically occurs at the Obr and Ti5c dual sites (i.e., H2 + Ti5c4+ + Obr2− → Ti5c4+-H− + ObrH−), the barrier decreases a lot to 0.58 eV. This indicates that the heterolytic mode is energetically more favorable for H-H bond scission than the homolytic mode. Moreover, the H atoms dissociated from the heterolytic cleavage would be eventually captured by Obr and Ti5c sites yielding a H+ and a H− anion, evidenced by their Bader charges of 1.00 |e| and −0.44 |e| (Fig. 1c), respectively. Notably, these barriers are slightly higher than those reported by Hu et al. (1.39 eV and 0.37 eV in homolytic and heterolytic modes, respectively), owing to the self-interaction error of TiO2 system ignored in their study [65].
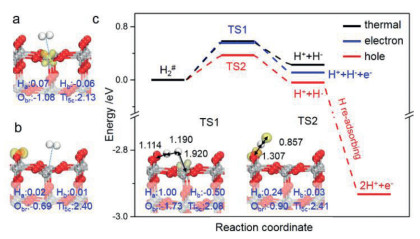
Secondly, we calculated the photocatalytic H2 dissociation on TiO2(110) surface. Generally, the generated photo-hole/electron pairs under light illumination thermodynamically tend to be trapped by Obr and Ti5c sites and form Obr·− and Ti5c3+, respectively, corresponding to Obr2− + h+ → Obr·− and Ti5c4+ + e– → Ti5c3+ [66-68]. As Figs. 2a and b show, our calculations verify that these two excited active sites (Obr·− and Ti5c3+) have insignificant effect on H2 adsorption with Ead(H2) of −0.22 eV and −0.21 eV, respectively. With the assistance of Ti5c3+, the barrier of H2 dissociation is nearly the same as that in thermocatalysis (0.56 eV vs. 0.58 eV) with a very similar TS structure (see TS1 in Fig. 2c), implying little contribution of photo-electron on H2 activation. Intriguingly, on hole-trapped TiO2(110) surface, the TS of H-H bond cleavage changes from the dihapto configuration in the thermocatalytic condition to the monohapto one (Figs. 2c and 1c), and the barrier is reduced to 0.37 eV with an enthalpy change of −0.04 eV (i.e., H2 + Obr·− → HObr− + H·). Notably, in comparison with the pristine homolytic cleavage mode in thermocatalysis, the barrier has largely declined (1.73 eV vs. 0.37 eV). With the generation of H· radical, it would further re-adsorb on Obr and form H+ with a large energy release (−2.90 eV). More specifically, in this photocatalytic TS, one H in H2 molecule is captured by Obr·− and the other H suspends above the TiO2 surface with the H···H and Ha···Obr bonds being 0.857 Å and 1.307 Å, respectively. In other word, it exhibits a radical-like TS, and the radical nature of H atom is also confirmed by Bader spin charge of 0.97 |e|. Therefore, the H-H bond activation mediated by photo-hole essentially follows the homolytic cleavage mechanism.
To shed light on the promotion effect of photo-hole on H2 dissociation, we quantitatively inspected the related electronic structure information. Firstly, the periodic natural bond orbital (NBO) [69] of two-site assisted TS complex in thermocatalysis was analyzed to uncover the bonding/antibonding nature of Ha-Obr and Hb-Ti5c (see Fig. 3a), in which Ha/Hb denotes the H atom in H2 close to the Obr/Ti5c site, respectively (Fig. 1a). Quantitatively, the charge of Ha is calculated to be 0.65 |e| in the H2 activation TS, indicating that the electron in Ha is partially shared by the forming Ha-Obr bond; however, it owns a charge of 1.06 |e| for the Hb atom in Hb-Ti5c bond, implying the negligible electron transfer (versus that (0.93 |e|) of Hb atom in pristine H2 molecule). Moreover, the occupancy of Ha-Obr bonding/antibonding is 0.91/0.20 |e|, but nearly zero for Hb-Ti5c bond. Therefore, we can speculate that the activation of H2 molecule depends mainly on the Obr site, whereas the Ti5c site contributes to a small extent, which could well explain the greater promotion effect of photo-hole than photo-electron. Similar result can be drawn in the C-H bond activation of CH4 molecule on rutile TiO2(110) surface [70].
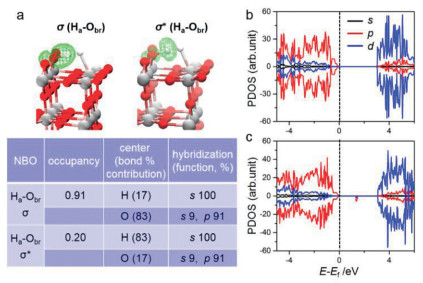
Furthermore, the projected densities of states (PDOSs) of TiO2(110) in the absence or presence of Obr·− were calculated (see Figs. 3b and c). It can be seen that the presence of localized photo-hole does not lead to significant change in the whole band gap, but induces a new unoccupied state within the forbidden band. In principle, this hole-state is conductive to accepting electron and exhibits strong oxidizability, evidenced by the higher energy level of Obr·− (~1.5 eV above the fermi level). In this sense, the superior performance of Obr·− than Obr2− on H-H bond rupture can be rationalized as follows. Obr·− holds less electron (−0.69 |e|) than Obr2− (−1.08 |e|), and this reactive Obr·− site, as a stronger oxidative center, can more easily accept electron from the dissociated hydrogen atom after H2 homolytic cleavage in photocatalysis, thereby largely decreasing the activation barrier.
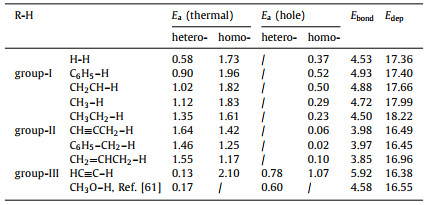
With the understanding of photoexcitation effect on modulating H2 dissociation, we are now at the position to explore the C-H bond activation in a series of different organic molecules on TiO2(110), aiming to uncover the general rule of the photo-hole effect, which includes (i) sp1–C group: HC≡C–H; (ii) sp2–C group: C6H5–H and CH2=CH–H; (iii) sp3–C group: CH3–H, CH3CH2–H, CH≡CCH2–H, C6H5CH2–H and CH2=CHCH2–H. At first, the thermally-driven C–H bond activation processes were studied. By comparing the barriers in homolytic and heterolytic cleavage modes (Table 1), we can see that heterolytic route is not always the most favorable one for all C-H bonds, which differs from H–H bond activation. In detail, the HC≡C-H, C6H5–H, CH2=CH–H, CH3–H and CH3CH2–H bonds prefer to be activated via the heterolytic cleavage mode, whereas the homolytic mode for the others (i.e., CH≡CCH2–H, C6H5CH2–H and CH2=CHCH2–H) (Fig. 4a and Table 1). Additionally, except for HC≡C–H (0.13 eV), almost all the reaction barriers of the most favored pathway are above 0.90 eV, which indicates the difficulty of C–H bond activation at room temperature. In the presence of Obr·−, we identified that the homolytic mode becomes more favored for C–H bonds in all but HC≡C–H (Table 1). Particularly, the activation barriers for the CH≡CCH2–H, C6H5CH2–H and CH2=CHCH2–H bonds are extremely small (< 0.10 eV) as compared to those in the thermocatalytic condition; for the usually-known inert C–H bonds in benzene, ethylene and methane, the barriers can also be reduced to below 0.52 eV. Overall, these results imply that for all these C–H bond in light hydrocarbons, the photo-hole can prompt their activation evidently, except for HC≡CH molecule.
 DownLoad:
CSV
DownLoad:
CSV
|
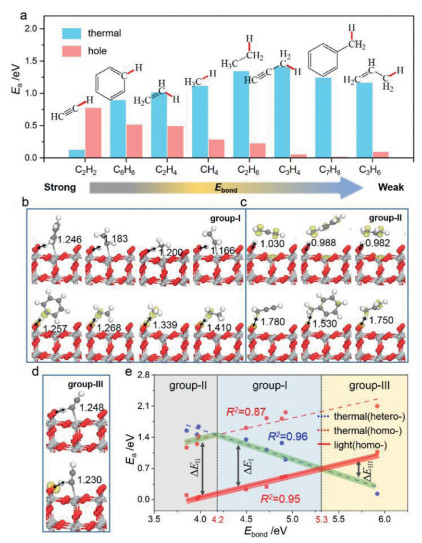
In addition, it is remarkable that the activation of these C–H bonds exhibits different behaviors, and is modulated by Obr·− to different degrees in comparison to the thermocatalysis (Fig. 4a). According to the results revealed above, these C–H bonds may be, therefore, classified as three groups: (group-Ⅰ) the ones (CH3–H, CH3CH2–H, CH2=CH–H and C6H5–H, Fig. 4b) that follow the heterolysis mode in thermocatalysis and change to the homolysis mode in photocatalysis, to which the photo-effect is beneficial (see ΔEI in Fig. 4e); (group-Ⅱ) the ones (CH≡CCH2–H, C6H5CH2–H and CH2=CHCH2–H, Fig. 4c) that follow the homolysis mode in thermocatalysis and keep it in photocatalysis, where the photo-effect is more efficient than group-Ⅰ (see ΔEⅡ in Fig. 4e); (group-Ⅲ) HC≡C–H (Fig. 4d), which prefers the heterolysis mode in both thermo- and photo-catalysis, and the presence of Obr·− will inhibit the HC≡C–H bond activation (see ΔEⅢ in Fig. 4e). One may naturally ask, what are the origins for these three different behaviors? Specifically, why do the group-Ⅱ C–H bonds tend to follow the homolytic cleavage mode, differing from the others? For HC≡C–H, why does it always follow the heterolysis mode, and why is the presence of Obr·− adverse to its C–H bond scission?
Toward these questions, firstly, we quantitatively tested the dependence of the Ea values via homolytic/heterolytic routes in the thermocatalytic condition on the corresponding C–H bond energies (Ebond). Two overall linear relations can be obtained with R2 of 0.87/0.96 (Fig. 4e), respectively. More intriguingly, as Ebond increases, Ea in the homolytic route increases, but decreases in the heterolytic one, and the crossing point of the two curves is at Ebond = 4.2 eV. These correlations clearly suggest that the choice of homolysis versus heterolysis in the thermocatalytic condition strongly depends on the C–H bond energy: the heterolysis is favored in the strong bond-energy region, while the homolysis is preferred in weak bond-energy region. For the group-Ⅱ C–H bonds, they own the relatively weaker Ebond among all the studied ones (Table 1), due to the stability of dehydrogenated molecule-fragment (i.e., CH≡C-CH2·, C6H5–CH2· and CH2=CH-CH2·) with extended π-electron conjugation, which rationalizes the preference of homolytic cleavage in thermocatalysis.
With the participation of Obr·− in the C–H bond activation, a similar linear correlation exists between the Ea following the homolysis mode in the photocatalytic condition and Ebond as well (R2 = 0.95, Fig. 4e). One can see from Fig. 4e that this line is overall downward shift in parallel as compared to that of the homolysis mode in the thermocatalytic condition, which again illustrates the promotion effect originating from the high oxidizability of Obr·−. Accordingly, we can explain the different promotion effect of Obr·− on the C–H bonds of group-Ⅰ versus group-Ⅱ, which gives ΔEⅡ > ΔEI. For group-Ⅱ C–H bonds, the homolytic mode is favored in both thermo- and photo-catalytic conditions, and the promotion extent (ΔEⅡ) of Obr·− is the largest as seen from Fig. 4e. Differently, for group-Ⅰ C–H bonds, the heterolysis mode is more favored in the thermocatalytic condition, and it will change to the homolysis mode in photocatalysis; thus, the photo-promotion effect (ΔEI) is relatively weakened relative to ΔEⅡ. Moreover, it is worth noting that ΔEI becomes smaller and smaller as the Ebond increases.
Secondly, as illustrated in Fig. 4e, with the increase of Ebond, we can see that the thermodynamically driven heterolytic mode becomes more and more feasible. When above ~5.3 eV, it will be even more favored than the photo-driven process, essentially ascribed to that the localized hole at Obr·− cannot provide enough energy to compensate the C–H bond energy. Specifically for HC≡C–H activation, the large Ebond of HC≡C–H is unfavorable for the occurring of homolytic mode. Moreover, as we know, HC≡CH together with the group-Ⅱ molecules (CH≡CCH3, C6H5-CH3 and CH2=CHCH3) displays weak acidity [72, 73], consistent with their relatively small deprotonation energies (Edep) in Table 1. These two factors imply that the HC≡C–H is more susceptible to releasing hydrogen in the form of proton (i.e., HC≡C–H → HC≡C− + H+). In this circumstance, the hydrogen in HC≡C–H would more easily combine with the Obr2− site via heterolysis rather than homolysis, which accordingly corresponds to a lower barrier of 0.13 eV (2.10 eV in homolysis). When the Obr·− is present, owing to the additional Coulomb repulsion between positively charged H+ and Obr·− (versus that in H+···Obr2−), the activation process is kinetically less favored than that in the pristine Obr2− (0.78 eV vs. 0.13 eV in photo- and thermo-catalysis, respectively). Thus, the HC≡C–H bond prefers to be activated through the heterolytic mode and photo-hole would suppress this activation process, as a result of its strong bond energy and acidity. Notably, the similar result was also observed in the heterolytic O–H bond cleavage in the photocatalytic CH3OH oxidation revealed in our previous study (Table 1) [61].
In summary, we have quantitatively studied the activation mechanism of H2 and a series of C–H bonds on TiO2(110) in the thermo- and photo-catalytic conditions, aiming to reveal the general photoexcitation effect on modulating the chemical bonds breakage. The main results can be summarized as follows:
(i) The thermal-driven H2 activation on TiO2(110) tends to obey the heterolytic cleavage mode, and the Obr·− radical formed from the photo-hole localization would change the mode to be homolytic one and evidently facilitate the activation process.
(ii) We identified that the examined C–H bonds can be classified as three groups in terms of the bond energy and acidity: group-Ⅰ (CH3–H, CH3CH2–H, CH2=CH–H and C6H5–H), group-Ⅱ (CH≡CCH2–H, C6H5CH2–H and CH2=CHCH2–H) and group-Ⅲ (HC≡C–H). The bond energy order is group-Ⅱ < group-Ⅰ < group-Ⅲ; meanwhile, group-Ⅱ/-Ⅲ C–H bonds own weak acidity. The Obr·− species can facilitate the activation of group-Ⅰ/Ⅱ C–H bonds to different degrees, whereas inhibits the one in HC≡C–H.
(iii) The different C–H activation behaviors in group-Ⅰ to group-Ⅲ could be largely attributed to the C–H bond energies and acidities. For the group-Ⅰ/-Ⅱ C–H bonds with relatively moderate/low bond energies, they obey the heterolytic/homolytic cleavage mechanism in thermocatalysis, and will uniformly obey the homolysis mechanism driven and simultaneously promoted by the highly oxidative Obr·−, where the promotion effect on group-Ⅱ C–H bonds is more evident with nearly negligible barriers.
(iv) Owing to the strong bond energy and weak acidity, we found that the HC≡C–H bond consistently prefers to be activated via the heterolytic cleavage mode, and Obr·− would inhibit this activation as a result of additional Coulomb repulsion between the dissociated proton and Obr·− (versus the pristine Obr2− on TiO2(110)).
This work provided a general atomic-level description on the photo- versus thermo-catalytic activation of H2 and various C–H bonds, which may explicitly deepen our understanding of the photocatalytic effect on modulating the chemical bonds breakage.
The authors report no declarations of interest.
This project was supported by National Nature Science Foundation of China (Nos. 21873028, 91945302), National Ten Thousand Talent Program for Young Top-notch Talents in China, Shanghai Shu-Guang project (No. 17SG30), and the Fundamental Research Funds for the Central Universities
R. Khorasani, P.E. Fleming, Comput. Theor. Chem. 1096 (2016) 89-93.
doi: 10.1016/j.comptc.2016.09.033
J. Berkowitz, G.B. Ellison, D. Gutman, J. Phys. Chem. 98 (1994) 2744-2765.
doi: 10.1021/j100062a009
B.B. Wayland, Polyhedron 7 (1988) 1545-1555.
doi: 10.1016/S0277-5387(00)81781-7
L. Meng, Z. Chen, Z. Ma, et al., Energy Environ. Sci. 11 (2018) 294-298.
doi: 10.1039/C7EE02951A
X. Li, W. Wang, F. Dong, et al., ACS Catal. 11 (2021) 4739-4769.
doi: 10.1021/acscatal.0c05354
M. Huš, D. Kopac, B. Likozar, J. Catal. 386 (2020) 126-138.
doi: 10.1016/j.jcat.2020.03.037
J.L. Gland, G.B. Fisher, E.B. Kollin, J. Catal. 77 (1982) 263-278.
doi: 10.1016/0021-9517(82)90167-1
G.E. Gdowski, R.J. Madix, Surf. Sci. 119 (1982) 184-206.
doi: 10.1016/0039-6028(82)90292-8
R. Van Lent, S.V. Auras, K. Cao, et al., Science 363 (2019) 155-157.
doi: 10.1126/science.aau6716
X. Qi, Y. Li, R. Bai, et al., Acc. Chem. Res. 50 (2017) 2799-2808.
doi: 10.1021/acs.accounts.7b00400
S.R. Neufeldt, G. JiméNez-Osés, J.R. Huckins, et al., J. Am. Chem. Soc. 137 (2015) 9843-9854.
doi: 10.1021/jacs.5b03535
J.C. Lewis, R.G. Bergman, J.A. Ellman, Acc. Chem. Res. 41 (2008) 1013-1025.
doi: 10.1021/ar800042p
M. Wijzenbroek, D. Helstone, J. Meyer, et al., J. Chem. Phys. 145 (2016) 144701.
doi: 10.1063/1.4964486
Q. Wu, L. Zhou, G.C. Schatz, et al., J. Am. Chem. Soc. 142 (2020) 13090-13101.
doi: 10.1021/jacs.0c04491
S. Mukherjee, F. Libisch, N. Large, et al., Nano Lett. 13 (2013) 240-247.
doi: 10.1021/nl303940z
T. Engel, H. Kuipers, Surf. Sci. 90 (1979) 181-196.
doi: 10.1016/0039-6028(79)90018-9
D.F. Padowitz, S.J. Sibener, Surf. Sci. 254 (1991) 125-143.
doi: 10.1016/0039-6028(91)90645-9
Y. Sun, S. Zhang, W. Zhang, et al., Chin. J. Chem. Phys. 31 (2018) 485-491.
doi: 10.1063/1674-0068/31/cjcp1805120
F. Zaera, D. Chrysostomou, Surf. Sci. 457 (2000) 89-108.
doi: 10.1016/S0039-6028(00)00337-X
M. L. Yang, Y.A. Zhu, C. Fan, et al., Phys. Chem. Chem. Phys. 13 (2011) 3257-3267.
doi: 10.1039/c0cp00341g
Y. Chen, D.G. Vlachos, J. Phys. Chem. C 114 (2010) 4973-4982.
W. Zhang, M. Pu, M. Lei, Langmuir 36 (2020) 5891-5901.
doi: 10.1021/acs.langmuir.0c00644
G. Righi, R. Magri, A. Selloni, J. Phys. Chem. C 123 (2019) 9875-9883.
doi: 10.1021/acs.jpcc.9b00609
L. Brugnoli, A. Pedone, M.C. Menziani, et al., J. Phys. Chem. C 123 (2019) 25668-25679.
doi: 10.1021/acs.jpcc.9b06805
Y. Chen, P. Hu, M.H. Lee, et al., Surf. Sci. 602 (2008) 1736-1741.
doi: 10.1016/j.susc.2008.02.036
Y. Chen, J. Cheng, P. Hu, et al., Surf. Sci. 602 (2008) 2828-2834.
doi: 10.1016/j.susc.2008.06.033
M.V. Bossche, H. Gronbeck, J. Am. Chem. Soc. 137 (2015) 12035-12044.
doi: 10.1021/jacs.5b06069
F. Zasada, J. Janas, W. Piskorz, et al., ACS Catal. 7 (2017) 2853-2867.
doi: 10.1021/acscatal.6b03139
Z. Zhu, W. Guo, Y. Zhang, et al., Carbon Energy 3 (2021) 519-540.
doi: 10.1002/cey2.127
T. Whittaker, K.B.S. Kumar, C. Peterson, et al., J. Am. Chem. Soc. 140 (2018) 16469-16487.
doi: 10.1021/jacs.8b04991
K. Sun, M. Kohyama, S. Tanaka, et al., J. Phys. Chem. C 118 (2014) 1611-1617.
doi: 10.1021/jp4099254
S. Mukherjee, L. Zhou, A.M. Goodman, et al., J. Am. Chem. Soc. 136 (2014) 64-67.
doi: 10.1021/ja411017b
N. Yodsin, C. Rungnim, S. Tungkamani, et al., J. Phys. Chem. C 124 (2019) 1941-1949.
Y. Chen, S. Ji, W. Sun, et al., Angew. Chem. Int. Ed. 132 (2020) 1295-1301.
doi: 10.1002/ange.201912043
H. She, H. Zhou, L. Li, et al., ACS Sustain. Chem. Eng. 6 (2018) 11939-11948.
doi: 10.1021/acssuschemeng.8b02217
T. Yan, Y. Wang, Y. Cao, et al., Appl. Catal. A: Gen. 630 (2022) 118457.
doi: 10.1016/j.apcata.2021.118457
C.F. Lien, M.T. Chen, Y.F. Lin, et al., J. Chin. Chem. Soc. 51 (2004) 37-42.
doi: 10.1002/jccs.200400007
H. Song, X. Meng, S. Wang, et al., J. Am. Chem. Soc. 141 (2019) 20507-20515.
doi: 10.1021/jacs.9b11440
X. Cao, T. Han, Q. Peng, et al., ChemComm 56 (2020) 13918-13932.
P.V.L. Reddy, K.H. Kim, H. Song, Renew. Sustain. Energy Rev. 24 (2013) 578-585.
doi: 10.1016/j.rser.2013.03.035
N. Feng, H. Lin, H. Song, et al., Nat. Commun. 12 (2021) 4652.
doi: 10.1038/s41467-021-24912-0
R. Sun, C. He, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 527-532.
doi: 10.1016/j.cclet.2021.05.072
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J. 414 (2021) 128857.
doi: 10.1016/j.cej.2021.128857
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149-3154.
doi: 10.1016/j.cclet.2021.02.046
Y. Zhao, W. Gao, S. Li, et al., Joule 3 (2019) 920-937.
doi: 10.1016/j.joule.2019.03.003
Y. Tian, L. Piao, X. Chen, Green Chem. 23 (2021) 3526-3541.
doi: 10.1039/D1GC00658D
W.B. Jiang, J.X. Low, et al., J. Univ. Sci. Technol. China 50 (2020) 1361.
M. Harb, G. Jeantelot, J.M. Basset, J. Phys. Chem. C 123 (2019) 28210-28218.
doi: 10.1021/acs.jpcc.9b08145
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169.
doi: 10.1103/PhysRevB.54.11169
G. Kresse, J. Furthmüller, Comput. Mater. Sci. 6 (1996) 15-50.
doi: 10.1016/0927-0256(96)00008-0
J.P. Perdew, A. Ruzsinszky, G.I. Csonka, Phys. Rev. Lett. 100 (2008) 136406.
doi: 10.1103/PhysRevLett.100.136406
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.
doi: 10.1103/PhysRevLett.77.3865
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758.
H. Yuan, H. Yang, P. Hu, et al., ACS Catal. 11 (2021) 6835-6845.
doi: 10.1021/acscatal.1c01050
H.F. Wang, D. Wang, X. Liu, et al., ACS Catal. 6 (2016) 5393-5398.
doi: 10.1021/acscatal.6b00764
H.F. Wang, R. Kavanagh, Y.L. Guo, et al., Angew. Chem. Int. Ed. 51 (2012) 6657-6661.
doi: 10.1002/anie.201108981
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799.
doi: 10.1002/jcc.20495
S. Grimme, J. Antony, S. Ehrlich, et al., J. Chem. Phys. 132 (2010) 154104.
doi: 10.1063/1.3382344
M. Setvin, C. Franchini, X. Hao, et al., Phys. Rev. Lett. 113 (2014) 086402.
doi: 10.1103/PhysRevLett.113.086402
D. Wang, H.F. Wang, P. Hu, Phys. Chem. Chem. Phys. 17 (2015) 1549-1555.
doi: 10.1039/C4CP04159C
J. Zhang, C. Peng, H. Wang, et al., ACS Catal. 7 (2017) 2374-2380.
doi: 10.1021/acscatal.6b03348
D. Wang, T. Sheng, J. Chen, et al., Nat. Catal. 1 (2018) 291-299.
doi: 10.1038/s41929-018-0055-z
H. Yuan, N. Sun, J. Chen, et al., ACS Catal. 8 (2018) 9269-9279.
doi: 10.1021/acscatal.8b02114
H. Yuan, J. Chen, H. Wang, et al., ACS Catal. 8 (2018) 10864-10870.
doi: 10.1021/acscatal.8b03045
G. Hu, Z. Wu, D.E. Jiang, J. Phys. Chem. C 122 (2018) 20323-20328.
doi: 10.1021/acs.jpcc.8b05251
J. Zhang, P. Zhou, J. Liu, et al., Phys. Chem. Chem. Phys. 16 (2014) 20382-20386.
doi: 10.1039/C4CP02201G
S. Wu, L. Wang, J. Zhang, J. Photochem. Photobiol. C Photochem. Rev. 46 (2021) 100400.
doi: 10.1016/j.jphotochemrev.2020.100400
H. Sheng, Q. Li, W. Ma, et al., Appl. Catal. B 138 (2013) 212-218.
B.D. Dunnington, J.R. Schmidt, J. Chem. Theory Comput. 8 (2012) 1902-1911.
doi: 10.1021/ct300002t
M. Zhou, H.F. Wang, JACS Au. 2 (2022) 188-196.
doi: 10.1021/jacsau.1c00466
P.J. Linstrom, W.G. Mallard, Chem. Eng. 46 (2001) 1059-1063.
P. Pässler, W. Hefner, K. Buckl, et al., Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000.
K.M. Ervin, S. Gronert, S.E. Barlow, et al., J. Am. Chem. Soc. 112 (1990) 5750-5759.
doi: 10.1021/ja00171a013
R. Khorasani, P.E. Fleming, Comput. Theor. Chem. 1096 (2016) 89-93.
doi: 10.1016/j.comptc.2016.09.033
J. Berkowitz, G.B. Ellison, D. Gutman, J. Phys. Chem. 98 (1994) 2744-2765.
doi: 10.1021/j100062a009
B.B. Wayland, Polyhedron 7 (1988) 1545-1555.
doi: 10.1016/S0277-5387(00)81781-7
L. Meng, Z. Chen, Z. Ma, et al., Energy Environ. Sci. 11 (2018) 294-298.
doi: 10.1039/C7EE02951A
X. Li, W. Wang, F. Dong, et al., ACS Catal. 11 (2021) 4739-4769.
doi: 10.1021/acscatal.0c05354
M. Huš, D. Kopac, B. Likozar, J. Catal. 386 (2020) 126-138.
doi: 10.1016/j.jcat.2020.03.037
J.L. Gland, G.B. Fisher, E.B. Kollin, J. Catal. 77 (1982) 263-278.
doi: 10.1016/0021-9517(82)90167-1
G.E. Gdowski, R.J. Madix, Surf. Sci. 119 (1982) 184-206.
doi: 10.1016/0039-6028(82)90292-8
R. Van Lent, S.V. Auras, K. Cao, et al., Science 363 (2019) 155-157.
doi: 10.1126/science.aau6716
X. Qi, Y. Li, R. Bai, et al., Acc. Chem. Res. 50 (2017) 2799-2808.
doi: 10.1021/acs.accounts.7b00400
S.R. Neufeldt, G. JiméNez-Osés, J.R. Huckins, et al., J. Am. Chem. Soc. 137 (2015) 9843-9854.
doi: 10.1021/jacs.5b03535
J.C. Lewis, R.G. Bergman, J.A. Ellman, Acc. Chem. Res. 41 (2008) 1013-1025.
doi: 10.1021/ar800042p
M. Wijzenbroek, D. Helstone, J. Meyer, et al., J. Chem. Phys. 145 (2016) 144701.
doi: 10.1063/1.4964486
Q. Wu, L. Zhou, G.C. Schatz, et al., J. Am. Chem. Soc. 142 (2020) 13090-13101.
doi: 10.1021/jacs.0c04491
S. Mukherjee, F. Libisch, N. Large, et al., Nano Lett. 13 (2013) 240-247.
doi: 10.1021/nl303940z
T. Engel, H. Kuipers, Surf. Sci. 90 (1979) 181-196.
doi: 10.1016/0039-6028(79)90018-9
D.F. Padowitz, S.J. Sibener, Surf. Sci. 254 (1991) 125-143.
doi: 10.1016/0039-6028(91)90645-9
Y. Sun, S. Zhang, W. Zhang, et al., Chin. J. Chem. Phys. 31 (2018) 485-491.
doi: 10.1063/1674-0068/31/cjcp1805120
F. Zaera, D. Chrysostomou, Surf. Sci. 457 (2000) 89-108.
doi: 10.1016/S0039-6028(00)00337-X
M. L. Yang, Y.A. Zhu, C. Fan, et al., Phys. Chem. Chem. Phys. 13 (2011) 3257-3267.
doi: 10.1039/c0cp00341g
Y. Chen, D.G. Vlachos, J. Phys. Chem. C 114 (2010) 4973-4982.
W. Zhang, M. Pu, M. Lei, Langmuir 36 (2020) 5891-5901.
doi: 10.1021/acs.langmuir.0c00644
G. Righi, R. Magri, A. Selloni, J. Phys. Chem. C 123 (2019) 9875-9883.
doi: 10.1021/acs.jpcc.9b00609
L. Brugnoli, A. Pedone, M.C. Menziani, et al., J. Phys. Chem. C 123 (2019) 25668-25679.
doi: 10.1021/acs.jpcc.9b06805
Y. Chen, P. Hu, M.H. Lee, et al., Surf. Sci. 602 (2008) 1736-1741.
doi: 10.1016/j.susc.2008.02.036
Y. Chen, J. Cheng, P. Hu, et al., Surf. Sci. 602 (2008) 2828-2834.
doi: 10.1016/j.susc.2008.06.033
M.V. Bossche, H. Gronbeck, J. Am. Chem. Soc. 137 (2015) 12035-12044.
doi: 10.1021/jacs.5b06069
F. Zasada, J. Janas, W. Piskorz, et al., ACS Catal. 7 (2017) 2853-2867.
doi: 10.1021/acscatal.6b03139
Z. Zhu, W. Guo, Y. Zhang, et al., Carbon Energy 3 (2021) 519-540.
doi: 10.1002/cey2.127
T. Whittaker, K.B.S. Kumar, C. Peterson, et al., J. Am. Chem. Soc. 140 (2018) 16469-16487.
doi: 10.1021/jacs.8b04991
K. Sun, M. Kohyama, S. Tanaka, et al., J. Phys. Chem. C 118 (2014) 1611-1617.
doi: 10.1021/jp4099254
S. Mukherjee, L. Zhou, A.M. Goodman, et al., J. Am. Chem. Soc. 136 (2014) 64-67.
doi: 10.1021/ja411017b
N. Yodsin, C. Rungnim, S. Tungkamani, et al., J. Phys. Chem. C 124 (2019) 1941-1949.
Y. Chen, S. Ji, W. Sun, et al., Angew. Chem. Int. Ed. 132 (2020) 1295-1301.
doi: 10.1002/ange.201912043
H. She, H. Zhou, L. Li, et al., ACS Sustain. Chem. Eng. 6 (2018) 11939-11948.
doi: 10.1021/acssuschemeng.8b02217
T. Yan, Y. Wang, Y. Cao, et al., Appl. Catal. A: Gen. 630 (2022) 118457.
doi: 10.1016/j.apcata.2021.118457
C.F. Lien, M.T. Chen, Y.F. Lin, et al., J. Chin. Chem. Soc. 51 (2004) 37-42.
doi: 10.1002/jccs.200400007
H. Song, X. Meng, S. Wang, et al., J. Am. Chem. Soc. 141 (2019) 20507-20515.
doi: 10.1021/jacs.9b11440
X. Cao, T. Han, Q. Peng, et al., ChemComm 56 (2020) 13918-13932.
P.V.L. Reddy, K.H. Kim, H. Song, Renew. Sustain. Energy Rev. 24 (2013) 578-585.
doi: 10.1016/j.rser.2013.03.035
N. Feng, H. Lin, H. Song, et al., Nat. Commun. 12 (2021) 4652.
doi: 10.1038/s41467-021-24912-0
R. Sun, C. He, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 527-532.
doi: 10.1016/j.cclet.2021.05.072
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J. 414 (2021) 128857.
doi: 10.1016/j.cej.2021.128857
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149-3154.
doi: 10.1016/j.cclet.2021.02.046
Y. Zhao, W. Gao, S. Li, et al., Joule 3 (2019) 920-937.
doi: 10.1016/j.joule.2019.03.003
Y. Tian, L. Piao, X. Chen, Green Chem. 23 (2021) 3526-3541.
doi: 10.1039/D1GC00658D
W.B. Jiang, J.X. Low, et al., J. Univ. Sci. Technol. China 50 (2020) 1361.
M. Harb, G. Jeantelot, J.M. Basset, J. Phys. Chem. C 123 (2019) 28210-28218.
doi: 10.1021/acs.jpcc.9b08145
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169.
doi: 10.1103/PhysRevB.54.11169
G. Kresse, J. Furthmüller, Comput. Mater. Sci. 6 (1996) 15-50.
doi: 10.1016/0927-0256(96)00008-0
J.P. Perdew, A. Ruzsinszky, G.I. Csonka, Phys. Rev. Lett. 100 (2008) 136406.
doi: 10.1103/PhysRevLett.100.136406
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.
doi: 10.1103/PhysRevLett.77.3865
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758.
H. Yuan, H. Yang, P. Hu, et al., ACS Catal. 11 (2021) 6835-6845.
doi: 10.1021/acscatal.1c01050
H.F. Wang, D. Wang, X. Liu, et al., ACS Catal. 6 (2016) 5393-5398.
doi: 10.1021/acscatal.6b00764
H.F. Wang, R. Kavanagh, Y.L. Guo, et al., Angew. Chem. Int. Ed. 51 (2012) 6657-6661.
doi: 10.1002/anie.201108981
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799.
doi: 10.1002/jcc.20495
S. Grimme, J. Antony, S. Ehrlich, et al., J. Chem. Phys. 132 (2010) 154104.
doi: 10.1063/1.3382344
M. Setvin, C. Franchini, X. Hao, et al., Phys. Rev. Lett. 113 (2014) 086402.
doi: 10.1103/PhysRevLett.113.086402
D. Wang, H.F. Wang, P. Hu, Phys. Chem. Chem. Phys. 17 (2015) 1549-1555.
doi: 10.1039/C4CP04159C
J. Zhang, C. Peng, H. Wang, et al., ACS Catal. 7 (2017) 2374-2380.
doi: 10.1021/acscatal.6b03348
D. Wang, T. Sheng, J. Chen, et al., Nat. Catal. 1 (2018) 291-299.
doi: 10.1038/s41929-018-0055-z
H. Yuan, N. Sun, J. Chen, et al., ACS Catal. 8 (2018) 9269-9279.
doi: 10.1021/acscatal.8b02114
H. Yuan, J. Chen, H. Wang, et al., ACS Catal. 8 (2018) 10864-10870.
doi: 10.1021/acscatal.8b03045
G. Hu, Z. Wu, D.E. Jiang, J. Phys. Chem. C 122 (2018) 20323-20328.
doi: 10.1021/acs.jpcc.8b05251
J. Zhang, P. Zhou, J. Liu, et al., Phys. Chem. Chem. Phys. 16 (2014) 20382-20386.
doi: 10.1039/C4CP02201G
S. Wu, L. Wang, J. Zhang, J. Photochem. Photobiol. C Photochem. Rev. 46 (2021) 100400.
doi: 10.1016/j.jphotochemrev.2020.100400
H. Sheng, Q. Li, W. Ma, et al., Appl. Catal. B 138 (2013) 212-218.
B.D. Dunnington, J.R. Schmidt, J. Chem. Theory Comput. 8 (2012) 1902-1911.
doi: 10.1021/ct300002t
M. Zhou, H.F. Wang, JACS Au. 2 (2022) 188-196.
doi: 10.1021/jacsau.1c00466
P.J. Linstrom, W.G. Mallard, Chem. Eng. 46 (2001) 1059-1063.
P. Pässler, W. Hefner, K. Buckl, et al., Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000.
K.M. Ervin, S. Gronert, S.E. Barlow, et al., J. Am. Chem. Soc. 112 (1990) 5750-5759.
doi: 10.1021/ja00171a013

Maosen Xu , Pengfei Zhu , Qinghong Cai , Meichun Bu , Chenghua Zhang , Hong Wu , Youzhou He , Min Fu , Siqi Li , Xingyan Liu . In-situ fabrication of TiO2/NH2−MIL-125(Ti) via MOF-driven strategy to promote efficient interfacial effects for enhancing photocatalytic NO removal activity. Chinese Chemical Letters, 2024, 35(10): 109524-. doi: 10.1016/j.cclet.2024.109524
Xiaoyao YIN , Wenhao ZHU , Puyao SHI , Zongsheng LI , Yichao WANG , Nengmin ZHU , Yang WANG , Weihai SUN . Fabrication of all-inorganic CsPbBr3 perovskite solar cells with SnCl2 interface modification. Chinese Journal of Inorganic Chemistry, 2025, 41(3): 469-479. doi: 10.11862/CJIC.20240309
Cailiang Yue , Nan Sun , Yixing Qiu , Linlin Zhu , Zhiling Du , Fuqiang Liu . A direct Z-scheme 0D α-Fe2O3/TiO2 heterojunction for enhanced photo-Fenton activity with low H2O2 consumption. Chinese Chemical Letters, 2024, 35(12): 109698-. doi: 10.1016/j.cclet.2024.109698
Huakang Zong , Xinyue Li , Yanlin Zhang , Faxun Wang , Xingxing Yu , Guotao Duan , Yuanyuan Luo . Pt/Ti3C2 electrode material used for H2S sensor with low detection limit and high stability. Chinese Chemical Letters, 2025, 36(5): 110195-. doi: 10.1016/j.cclet.2024.110195
Hongye Bai , Lihao Yu , Jinfu Xu , Xuliang Pang , Yajie Bai , Jianguo Cui , Weiqiang Fan . Controllable Decoration of Ni-MOF on TiO2: Understanding the Role of Coordination State on Photoelectrochemical Performance. Chinese Journal of Structural Chemistry, 2023, 42(10): 100096-100096. doi: 10.1016/j.cjsc.2023.100096
Qiuyun Li , Yannan Zhu , Yining Wang , Gang Qi , Wen-Juan Hao , Kelu Yan , Bo Jiang . Catalytic CH activation-initiated transdiannulation: An oxygen transfer route to ring-fluorinated tricyclic γ-lactones. Chinese Chemical Letters, 2024, 35(9): 109494-. doi: 10.1016/j.cclet.2024.109494
Zhiqiang Wang , Yajie Gao , Tianjun Wang , Wei Chen , Zefeng Ren , Xueming Yang , Chuanyao Zhou . Photocatalyzed oxidation of water on oxygen pretreated rutile TiO2(110). Chinese Chemical Letters, 2025, 36(4): 110602-. doi: 10.1016/j.cclet.2024.110602
Yue Zhang , Xiaoya Fan , Xun He , Tingyu Yan , Yongchao Yao , Dongdong Zheng , Jingxiang Zhao , Qinghai Cai , Qian Liu , Luming Li , Wei Chu , Shengjun Sun , Xuping Sun . Ambient electrosynthesis of urea from carbon dioxide and nitrate over Mo2C nanosheet. Chinese Chemical Letters, 2024, 35(8): 109806-. doi: 10.1016/j.cclet.2024.109806
Jiangqi Ning , Junhan Huang , Yuhang Liu , Yanlei Chen , Qing Niu , Qingqing Lin , Yajun He , Zheyuan Liu , Yan Yu , Liuyi Li . Alkyl-linked TiO2@COF heterostructure facilitating photocatalytic CO2 reduction by targeted electron transport. Chinese Journal of Structural Chemistry, 2024, 43(12): 100453-100453. doi: 10.1016/j.cjsc.2024.100453
Jiatong Li , Linlin Zhang , Peng Huang , Chengjun Ge . Carbon bridge effects regulate TiO2–acrylate fluoroboron coatings for efficient marine antifouling. Chinese Chemical Letters, 2025, 36(2): 109970-. doi: 10.1016/j.cclet.2024.109970
Zhen Shi , Wei Jin , Yuhang Sun , Xu Li , Liang Mao , Xiaoyan Cai , Zaizhu Lou . Interface charge separation in Cu2CoSnS4/ZnIn2S4 heterojunction for boosting photocatalytic hydrogen production. Chinese Journal of Structural Chemistry, 2023, 42(12): 100201-100201. doi: 10.1016/j.cjsc.2023.100201
Ziruo Zhou , Wenyu Guo , Tingyu Yang , Dandan Zheng , Yuanxing Fang , Xiahui Lin , Yidong Hou , Guigang Zhang , Sibo Wang . Defect and nanostructure engineering of polymeric carbon nitride for visible-light-driven CO2 reduction. Chinese Journal of Structural Chemistry, 2024, 43(3): 100245-100245. doi: 10.1016/j.cjsc.2024.100245
Mengjun Zhao , Yuhao Guo , Na Li , Tingjiang Yan . Deciphering the structural evolution and real active ingredients of iron oxides in photocatalytic CO2 hydrogenation. Chinese Journal of Structural Chemistry, 2024, 43(8): 100348-100348. doi: 10.1016/j.cjsc.2024.100348
Jiaqi Ma , Lan Li , Yiming Zhang , Jinjie Qian , Xusheng Wang . Covalent organic frameworks: Synthesis, structures, characterizations and progress of photocatalytic reduction of CO2. Chinese Journal of Structural Chemistry, 2024, 43(12): 100466-100466. doi: 10.1016/j.cjsc.2024.100466
Weixu Li , Yuexin Wang , Lin Li , Xinyi Huang , Mengdi Liu , Bo Gui , Xianjun Lang , Cheng Wang . Promoting energy transfer pathway in porphyrin-based sp2 carbon-conjugated covalent organic frameworks for selective photocatalytic oxidation of sulfide. Chinese Journal of Structural Chemistry, 2024, 43(7): 100299-100299. doi: 10.1016/j.cjsc.2024.100299
Xinyue Han , Yunhan Yang , Jiayin Lu , Yuxiang Lin , Dongxue Zhang , Ling Lin , Liang Qiao . Efficient serum lipids profiling by TiO2-dopamin-assisted MALDI-TOF MS for breast cancer detection. Chinese Chemical Letters, 2025, 36(5): 110183-. doi: 10.1016/j.cclet.2024.110183
Zhijia Zhang , Shihao Sun , Yuefang Chen , Yanhao Wei , Mengmeng Zhang , Chunsheng Li , Yan Sun , Shaofei Zhang , Yong Jiang . Epitaxial growth of Cu2-xSe on Cu (220) crystal plane as high property anode for sodium storage. Chinese Chemical Letters, 2024, 35(7): 108922-. doi: 10.1016/j.cclet.2023.108922
Ting Xie , Xun He , Lang He , Kai Dong , Yongchao Yao , Zhengwei Cai , Xuwei Liu , Xiaoya Fan , Tengyue Li , Dongdong Zheng , Shengjun Sun , Luming Li , Wei Chu , Asmaa Farouk , Mohamed S. Hamdy , Chenggang Xu , Qingquan Kong , Xuping Sun . CoSe2 nanowire array enabled highly efficient electrocatalytic reduction of nitrate for ammonia synthesis. Chinese Chemical Letters, 2024, 35(11): 110005-. doi: 10.1016/j.cclet.2024.110005
Yanghanbin Zhang , Dongxiao Wen , Wei Sun , Jiahe Peng , Dezhong Yu , Xin Li , Yang Qu , Jizhou Jiang . State-of-the-art evolution of g-C3N4-based photocatalytic applications: A critical review. Chinese Journal of Structural Chemistry, 2024, 43(12): 100469-100469. doi: 10.1016/j.cjsc.2024.100469
Guixu Pan , Zhiling Xia , Ning Wang , Hejia Sun , Zhaoqi Guo , Yunfeng Li , Xin Li . Preparation of high-efficient donor-π-acceptor system with crystalline g-C3N4 as charge transfer module for enhanced photocatalytic hydrogen evolution. Chinese Journal of Structural Chemistry, 2024, 43(12): 100463-100463. doi: 10.1016/j.cjsc.2024.100463
 DownLoad:
DownLoad: