
 Login In
Login InSynthesis and in vitro antitumor activity of novel naphthyridinone derivatives
Figures(3)
Citation:
. Synthesis and in vitro antitumor activity of novel naphthyridinone derivatives[J]. Chinese Chemical Letters,
;2017, 28(2): 235-239.
doi:
10.1016/j.cclet.2016.07.024

Figures(3)




Cancers still remain the first and second leading causes of death in economically developed and developing countries, respectively [1, 2]. It is expected that annual cancer cases will rise from 14 million in 2012 to 22 million within the next two decades [3]. Encouragingly, substantial progress has been made to develop more effective and safer strategies for treating cancers, and many classes of antitumor agents with diverse novel structures have been introduced in the market or under development in the past decades [4]. However, there continues to be a need for new antitumor drugs from either novel scaffolds or further structural modifications of existing drugs.
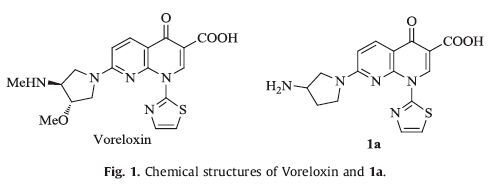
Quinolones represent an extremely successful family of antibiotics that have a broad-spectrum antibacterial activity and relatively few side effects [5]. Among of them, ciprofloxacin, ofloxacin and levofloxacin are also used as second-line drugs to treat tuberculosis [6]. Recently, quinolones as "privileged building blocks" have been reported to display many "nonclassical" biological profiles, such as antitumor, anti-HIV-1 integrase, anti-HCV-NS3 helicase and -NS5B-polymerase activities [7, 8]. It's exciting that Voreloxin (Fig. 1) with a broad-spectrum antitumor activity [9] the first quinolone antitumor drug, was approved as an orphan drug for the treatment of acute myeloid leukemia by the US FDA in 2009, and phase I-III clinical trials in patients with various tumors are currently ongoing [10].
In the course of our search for more potent antitumor agents, we focused our interest on structural modifications of Voreloxin. Given that 3-aminopyrrolidine, but not (3S, 4S)-3-methoxy-4-(methylamino) pyrrolidine, is commercially available, compound 1a (Fig. 1) possessing the in vitro antitumor activity comparable to Voreloxin [11] was chosen as the lead compound in our work. First, the thiazoly-2-yl group at the N-1 position of 1a was replaced with its isosteres (1H-imidazol-2-yl and oxazol-2-yl) to examine whether it is the optimal group of this position. On the other hand, our previous works have emphasized the importance of an oxime functional moiety of the C-7 side chain with respect to biological activities of quinolones [12-15]. Thus, a four, five-or sixmembered nitrogen heterocycle containing an alkoxyimino group was introduced at the C-7 position instead of the 3-aminopyrrolidin-1-yl one of 1a, to identify if a nitrogen heterocycle with an oxime moiety is permitted at this position. Our primary objective was to optimize the potency of these compounds against human cancer cell lines. A simple structure-activity relationship (SAR) study was also explored to facilitate the further development of the naphthyridinone derivatives.
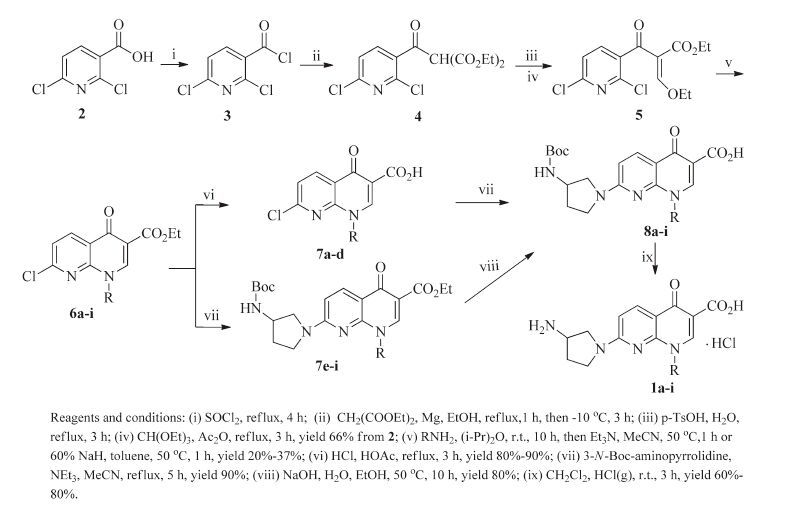
To study the effect of various substituents at the N-1 position of the 7-(3-aminopyrrolidin-1-yl) naphthyridinone core, eight analogs of 1a were designed. A synthetic route to these compounds is illustrated in Scheme 1. The key intermediates 6a-i were readily prepared from 2, 6-dichloronicotinic acid (2) via a five-step procedure [14, 16]. Hydrolysis of the core esters 6a-d in HCl-HOAc followed by condensation with 3-N-Boc-aminopyrrolidine in the presence of triethylamine gave 8a-d. However, the esters 6e-i were not converted to the corresponding acids in a similar manner as for preparation of 8a-d. After various attempts, 8e-i were successfully obtained from 6e-i by condensation with the side chain compounds and then hydrolysis of the resulted esters 7e-i. The Boc-protecting group of 8a-i was removed by hydrogen chloride gas in dichloromethane to yield the desired naphthyridinone derivatives 1a-i as hydrochloric acid salts.
The synthetic route of novel 1-(thiazol-2-yl) naphthyridinones 10a-n containing nitrogen heterocycles with an alkoxyimino group was depicted in Scheme 2. Condensation of 6a with various side chain compounds [12, 17-19] and then hydrolysis of the resulted esters 9a-n yielded 10a-n.
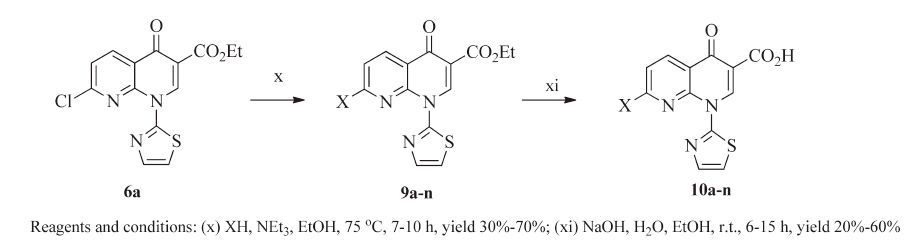
Since the oxime group can exist in the E or Z configuration, it was necessary to determine the geometries of all the oxime target compounds 10a-n. Preparing X-ray quality single crystals of any oxime intermediate or product met with no success in this study, but the oxime geometry would be expected to have the E-configuration according to the data in published papers [20, 21]. The concrete synthetic procedures, the physical characteristics, and 1H NMR for all the synthesized compounds are listed in Supporting information.
All the synthesized target compounds were preliminarily investigated for their in vitro activity against HL60 (leukemia) at the concentration of 30 μmol/L by SRB (Sulforhodamine B) assay [22]. And the compounds having >70% inhibition were subjected to IC50 (50% inhibition concentrations) determination against ten human cancer cell lines, including HL60, HepG2 (liver carcinoma), HCT-116 (colon cancer), A549 (lung adenocarcinoma), PANC-1 (pancreatic carcinoma), Hela (cervical cancer), DU145 (prostatic cancer), SKOV3 (ovarian carcinoma), MCF-7 (breast cancer) and MCF-7/DOX (Doxorubicin-resistant MCF-7) by SRB assay.
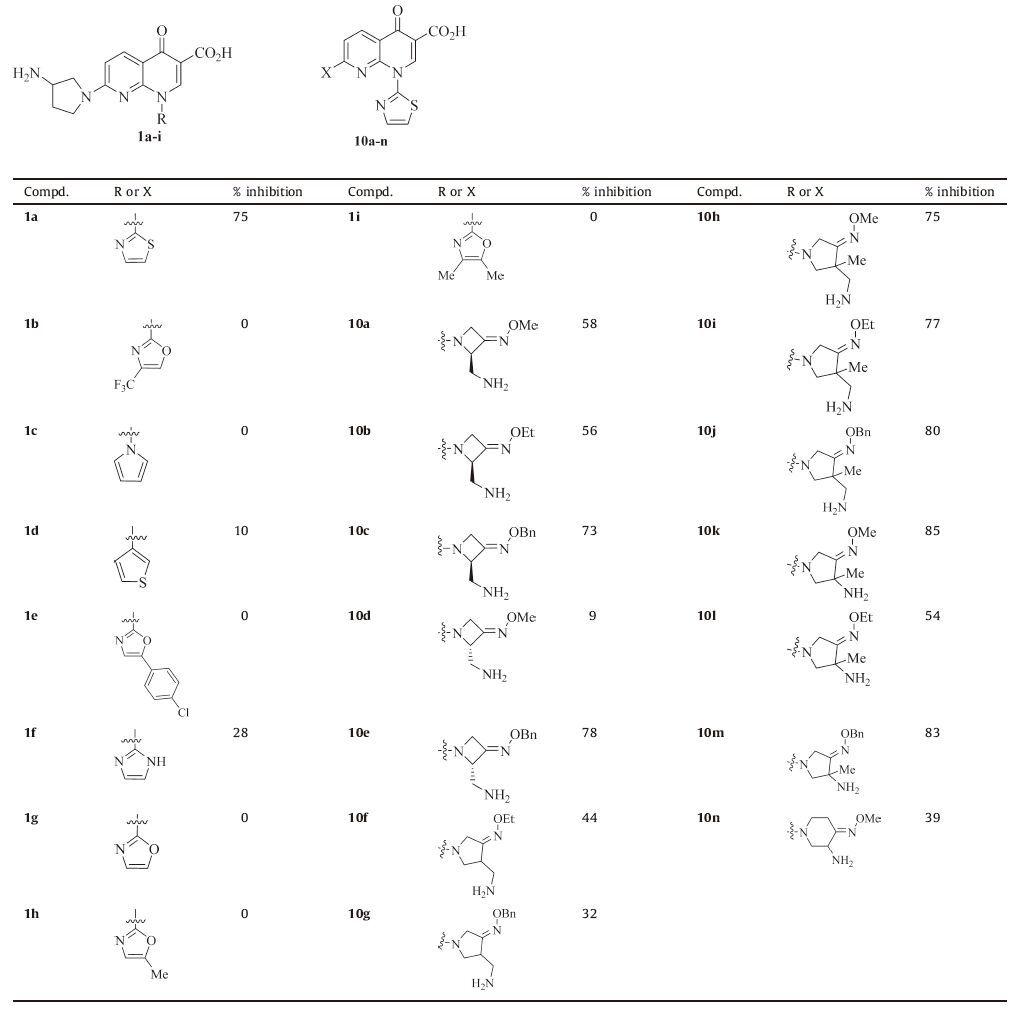
The inhibitory effects of the 7-(3-aminopyrrolidin-1-yl) naphthyridinone derivatives 1a-i and 1-(thiazol-2-yl) naphthyridinone derivatives 10a-n were evaluated and listed in Tables 1 and 2. Unfortunately, contrary to our predictions, the results indicated that all the newly synthesized 7-(3-aminopyrrolidin-1-yl) naphthyridinone derivatives 1b-i (inhibition rates: 0-28%) were much less active than the reference 1a (75%, Table 1). Therefore, our modifications are not effective, and the thiazol-2-yl group is optimal for the N-1 position.
 下载:
导出CSV
下载:
导出CSV
 |
下载:
导出CSV
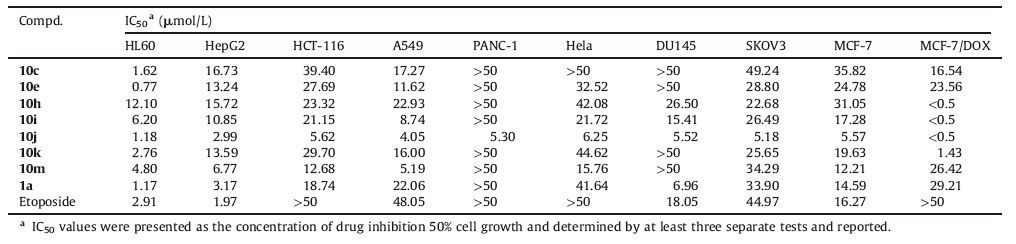 |
The IC50 values were compared with those of Etoposide and 1a (Table 2). The selected 1-(thiazol-2-yl) naphthyridinones have potent activity against these tested human cancer cell lines. All of them (IC50: < 0.5-49.23 μmol/L) are more active or comparable to Etoposide and 1a (IC50: 1.17 to >50 μmol/L) against HCT-116, A549, SKOV3 and MCF-7/DOX. Contrary to 1a, compounds 10h-j show significantly better activity against MCF-7/DOX (IC50: < 0.5 μmol/L) than MCF-7 (IC50: 5.57-31.04 μmol/L). Moreover, 10j was found to have a broad-spectrum activity (IC50: < 0.5-6.25 μmol/L) against all of the tested cell lines including Etoposide-and/or 1a -resistant ones, and is 1.3 to >100 fold more potent than those of the two references against these cell lines, except HL60 and HepG2.
According to the biological evaluation results shown in Tables 1 and 2, the antitumor activity of the naphthyridinone derivatives in this study depends on both of the groups at the N-1 and C-7 positions. The relative contribution of the heteroaromatic ring at the N-1 position to activity is as follows: thiazol-2-yl (1a) >> 1H-imidazol-2-yl (1f) >thiophen-3-yl (1d), and the others (1b, 1c, 1e, 1g-i) have no activity (Table 1).
On the other hand, 1-(thiazol-2-yl) naphthyridinones 10a-n generally exhibit in vitro activity (Table 1), which indicates that a nitrogen heterocycle with an oxime group is permitted at the C-7 position. Although a simple SAR is difficult to explore, the sizes of the heterocycle and the oxime group are important for the activity. For example, the activity imparted to the 7-(3-aminomethyl-3-methylpyrrolidin-1-yl) naphthyridinone ring by the alkyl group of the oxime moiety is as follows: benzyl > ethyl > methyl (10j vs.10i vs.10h), which suggests that simply increasing the lipophilicity could improve the activity (Table 2). Above all, thiazol-2-yl and 3-aminomethyl-4-benzyl oxyimino-3-methylpyrrolidin-1-yl groups are the most active at the N-1 and C-7 positions of naphthyridinone core, respectively.
In summary, structural modifications of compound 1a (a precursor of Voreloxin) at the N-1 position (various heteroaromatic rings instead of the thiazol-2-yl) and C-7 position (four-/ five-/six-membered nitrogen heterocyclic amine moieties with an oxime group instead of 3-aminopyrrolidin-1-yl one), respectively, were made in this study. Our results reveal that thiazol-2-yl and 3-aminomethyl-4-benzyloxyimino-3-methylpyrrolidin-1-yl groups are optimal at the N-1 and C-7 positions of naphthyridinone core, respectively. The most active compound 10j shows considerable broad-spectrum antitumor activity (IC50: < 0.5-6.25 μmol/L) against all of the tested cell lines including Etoposide-and/or 1a-resistant ones and is 1.3 to >100 fold more potent than those of the two references against these cell lines, except HL60 and HepG2.
This work was supported by National Key Research and Development Program (No. 2016YFA0201500), the National S & T Major Special Project on Major New Drug Innovations (Nos. 2014ZX09507009-003, 2015ZX09102007) and NSFC (Nos. 81373267, 21502237).
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.07.024.

Jian Song , Shenghui Wang , Qiuge Liu , Xiao Wang , Shuo Yuan , Hongmin Liu , Saiyang Zhang . N-Benzyl arylamide derivatives as novel and potent tubulin polymerization inhibitors against gastric cancers: Design, structure–activity relationships and biological evaluations. Chinese Chemical Letters, 2025, 36(2): 109678-. doi: 10.1016/j.cclet.2024.109678
Shushan Mo , Zhaoshuo Wang , Dandan Ding , Zhengzheng Yan , Yunlu Dai , Jinchao Zhang , Huifang Liu , Tianjiao Liang , Jianfei Tong , Zhenhua Li , Xueyi Wang . The synthesis and evaluation of novel BPA derivatives for enhanced blood-brain barrier penetration and boron neutron capture therapy. Chinese Chemical Letters, 2025, 36(5): 110190-. doi: 10.1016/j.cclet.2024.110190
Bairu Meng , Zongji Zhuo , Han Yu , Sining Tao , Zixuan Chen , Erik De Clercq , Christophe Pannecouque , Dongwei Kang , Peng Zhan , Xinyong Liu . Design, synthesis, and biological evaluation of benzo[4,5]thieno[2,3-d]pyrimidine derivatives as novel HIV-1 NNRTIs. Chinese Chemical Letters, 2024, 35(6): 108827-. doi: 10.1016/j.cclet.2023.108827
Bofei JIA , Zhihao LIU , Zongyuan GAO , Shuai ZHOU , Mengxiang WU , Qian ZHANG , Xiamei ZHANG , Shuzhong CHEN , Xiaohan YANG , Yahong LI . Cu(Ⅱ) and Cu(Ⅰ) complexes based on derivatives of imidazo[1,5-a]pyridine: Synthesis, structures, in situ metal-ligand reactions, and catalytic activity. Chinese Journal of Inorganic Chemistry, 2025, 41(5): 1020-1036. doi: 10.11862/CJIC.20240317
Di ZHANG , Tianxiang XIE , Xu HE , Wanyu WEI , Qi FAN , Jie QIAO , Gang JIN , Ningbo LI . Construction and antitumor activity of pH/GSH dual-responsive magnetic nanodrug. Chinese Journal of Inorganic Chemistry, 2025, 41(4): 786-796. doi: 10.11862/CJIC.20240329
Fengqing Wang , Changxing Qi , Chunmei Chen , Qin Li , Qingyi Tong , Weiguang Sun , Zhengxi Hu , Minyan Wang , Hucheng Zhu , Lianghu Gu , Yonghui Zhang . Discovery and enantioselective total synthesis of antitumor agent asperfilasin A via a regio- and diastereoselective Nazarov cyclization. Chinese Chemical Letters, 2025, 36(6): 110252-. doi: 10.1016/j.cclet.2024.110252
Liping Zhao , Xixi Guo , Zhimeng Zhang , Xi Lu , Qingxuan Zeng , Tianyun Fan , Xintong Zhang , Fenbei Chen , Mengyi Xu , Min Yuan , Zhenjun Li , Jiandong Jiang , Jing Pang , Xuefu You , Yanxiang Wang , Danqing Song . Novel berberine derivatives as adjuvants in the battle against Acinetobacter baumannii: A promising strategy for combating multi-drug resistance. Chinese Chemical Letters, 2024, 35(10): 109506-. doi: 10.1016/j.cclet.2024.109506
Wenyi Mei , Lijuan Xie , Xiaodong Zhang , Cunjian Shi , Fengzhi Wang , Qiqi Fu , Zhenjiang Zhao , Honglin Li , Yufang Xu , Zhuo Chen . Design, synthesis and biological evaluation of fluorescent derivatives of ursolic acid in living cells. Chinese Chemical Letters, 2024, 35(5): 108825-. doi: 10.1016/j.cclet.2023.108825
Hong-Tao Ji , Yu-Han Lu , Yan-Ting Liu , Yu-Lin Huang , Jiang-Feng Tian , Feng Liu , Yan-Yan Zeng , Hai-Yan Yang , Yong-Hong Zhang , Wei-Min He . Nd@C3N4-photoredox/chlorine dual catalyzed synthesis and evaluation of antitumor activities of 4-alkylated sulfonyl ketimines. Chinese Chemical Letters, 2025, 36(2): 110568-. doi: 10.1016/j.cclet.2024.110568
Guo-Ping Yin , Ya-Juan Li , Li Zhang , Ling-Gao Zeng , Xue-Mei Liu , Chang-Hua Hu . Citrinsorbicillin A, a novel homotrimeric sorbicillinoid isolated by LC-MS-guided with cytotoxic activity from the fungus Trichoderma citrinoviride HT-9. Chinese Chemical Letters, 2024, 35(8): 109035-. doi: 10.1016/j.cclet.2023.109035
Shuying Li , Weiwei ZhuGe , Xuan Sun , Chongzhen Sun , Zhaojun Liu , Chenghe Xiong , Min Xiao , Guofeng Gu . Convergent synthesis and immunological study of oligosaccharide derivatives related to galactomannan from Antrodia cinnamomea. Chinese Chemical Letters, 2024, 35(5): 109089-. doi: 10.1016/j.cclet.2023.109089
Yulong Shi , Fenbei Chen , Mengyuan Wu , Xin Zhang , Runze Meng , Kun Wang , Yan Wang , Yuheng Mei , Qionglu Duan , Yinghong Li , Rongmei Gao , Yuhuan Li , Hongbin Deng , Jiandong Jiang , Yanxiang Wang , Danqing Song . Chemical construction and anti-HCoV-OC43 evaluation of novel 10,12-disubstituted aloperine derivatives as dual cofactor inhibitors of TMPRSS2 and SR-B1. Chinese Chemical Letters, 2024, 35(5): 108792-. doi: 10.1016/j.cclet.2023.108792
Anqiu LIU , Long LIN , Dezhi ZHANG , Junyu LEI , Kefeng WANG , Wei ZHANG , Junpeng ZHUANG , Haijun HAO . Synthesis, structures, and catalytic activity of aluminum and zinc complexes chelated by 2-((2,6-dimethylphenyl)amino)ethanolate. Chinese Journal of Inorganic Chemistry, 2024, 40(4): 791-798. doi: 10.11862/CJIC.20230424
Guoping Yang , Zhoufu Lin , Xize Zhang , Jiawei Cao , Xuejiao Chen , Yufeng Liu , Xiaoling Lin , Ke Li . Assembly of Y(Ⅲ)-containing antimonotungstates induced by malic acid with catalytic activity for the synthesis of imidazoles. Chinese Chemical Letters, 2024, 35(12): 110274-. doi: 10.1016/j.cclet.2024.110274
Yao HUANG , Yingshu WU , Zhichun BAO , Yue HUANG , Shangfeng TANG , Ruixue LIU , Yancheng LIU , Hong LIANG . Copper complexes of anthrahydrazone bearing pyridyl side chain: Synthesis, crystal structure, anticancer activity, and DNA binding. Chinese Journal of Inorganic Chemistry, 2025, 41(1): 213-224. doi: 10.11862/CJIC.20240359
Zhiwei Chen , Heyun Sheng , Xue Li , Menghan Chen , Xin Li , Qiuling Song . Efficient capture of difluorocarbene by pyridinium 1,4-zwitterionic thiolates: A concise synthesis of difluoromethylene-containing 1,4-thiazine derivatives. Chinese Chemical Letters, 2024, 35(4): 108937-. doi: 10.1016/j.cclet.2023.108937
Maitri Bhattacharjee , Rekha Boruah Smriti , R. N. Dutta Purkayastha , Waldemar Maniukiewicz , Shubhamoy Chowdhury , Debasish Maiti , Tamanna Akhtar . Synthesis, structural characterization, bio-activity, and density functional theory calculation on Cu(Ⅱ) complexes with hydrazone-based Schiff base ligands. Chinese Journal of Inorganic Chemistry, 2024, 40(7): 1409-1422. doi: 10.11862/CJIC.20240007
Xiaomeng Hu , Jie Yu , Lijie Sun , Linfeng Zhang , Wei Zhou , Dongpeng Yan , Xinrui Wang . Synthesis of an AVB@ZnTi-LDH composite with synergistically enhance UV blocking activity and high stability for potential application in sunscreen formulations. Chinese Chemical Letters, 2024, 35(11): 109466-. doi: 10.1016/j.cclet.2023.109466
Ao Sun , Zipeng Li , Shuchun Li , Xiangbao Meng , Zhongtang Li , Zhongjun Li . Stereoselective synthesis of α-3-deoxy-D-manno-oct-2-ulosonic acid (α-Kdo) derivatives using a C3-p-tolylthio-substituted Kdo fluoride donor. Chinese Chemical Letters, 2025, 36(3): 109972-. doi: 10.1016/j.cclet.2024.109972
Ping Sun , Yuanqin Huang , Shunhong Chen , Xining Ma , Zhaokai Yang , Jian Wu . Indole derivatives as agrochemicals: An overview. Chinese Chemical Letters, 2024, 35(7): 109005-. doi: 10.1016/j.cclet.2023.109005
 DownLoad:
DownLoad: