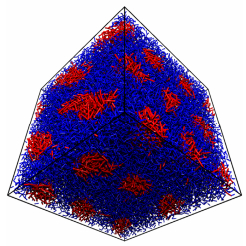
 Figure 1.
The sphere structure of R5C30R5 triblock copolymers in bulk Red and blue lines represent monomers R and C, respectively. Symbols are the same in the remaining figures. System size Lx = Ly = Lz = 30 and PBCs along all directions.
Figure 1.
The sphere structure of R5C30R5 triblock copolymers in bulk Red and blue lines represent monomers R and C, respectively. Symbols are the same in the remaining figures. System size Lx = Ly = Lz = 30 and PBCs along all directions.
Citation:
Ya-juan Su, Jian-hua Huang. Self-assembly Behavior of Rod-Coil-Rod Triblock Copolymers within a Planar Slit[J]. Chinese Journal of Polymer Science,
2016, 34(7): 838-849.
doi:
10.1007/s10118-016-1803-7

Self-assembly Behavior of Rod-Coil-Rod Triblock Copolymers within a Planar Slit
English
Self-assembly Behavior of Rod-Coil-Rod Triblock Copolymers within a Planar Slit
Abstract:
The self-assembly behavior of sphere-forming R5C30R5 triblock copolymers within a planar slit is studied by performing dissipative particle dynamics simulations. A sequence of novel structures which are not observed in bulk are formed within slits, including wetting layers, island-like structure, parallel cylinders, perpendicular cylinders and cross-cylindrical structures. Perpendicular cylinders are always formed before the increase in the layers of parallel cylinders. A phase diagram of the assembled structures with respective to the slit property and height is thus presented. The rod length is found to have a significant impact on the rod alignment, and a disordered-ordered transition of rod orientation occurs with an increase in the length of rod blocks. Some special structures, such as parallel half-cylinders and arrowhead-shaped morphology, are observed when the rod length increases to a certain extent. Our results show that the property and height of the slit and rod length all influence the self-assembly of rod-coil-rod triblock copolymers.
-
Key words:
- Self-assembly
- / Triblock copolymers
- / Slit
- / Simulation
-
INTRODUCTION
Block copolymers (BCPs) are known to self-assemble into various ordered microstructures[1, 2]. For BCPs in bulk, the ordered phases include lamellae (L), gyroid (G), hexagonally packed cylinders (C), and a body-centered-cubic array of spheres (S) dependent on the copolymer composition[3, 4]. Recently, the self-assembly of confined BCPs has received considerable attention because of its potential applications in nanofabrication[5]. Such potential applications are emerging in various areas, such as high density data storage, nanolithography, photonic crystals and so on. Phase behavior of BCPs in films is more complex than that in bulk. The confinement and the surface field induce BCPs to form different orientations and microstructures from the bulk[6]. It is important to control the orientation of the spatial structure over rather large length scales for many practical applications. Moreover, BCP films seem to be ideal systems for studying the competition among chain stretching, interfacial tension, and external field, which, in turn, allows the selection of ordered morphologies[7].
Understanding the assembly of confined block copolymers has been an active research area in recent years. Compared with the classic flexible block copolymers, copolymers which contain rod-like blocks exhibit unique phase behavior due to the tendency of liquid crystalline or crystalline ordering of the rodlike chain[8, 9]. However, previous studies on the confinement system mostly focused on rod(R)-coil(C) BCPs which contain only one rigid or rodlike block[10-15].Smectic RC melts confined within relatively thick films formed well ordered lamellae either parallel or perpendicular to the surfaces through the change in the rod tilt angle adjacent to the surfaces[10]. By using self-consistent field theory (SCFT), Yang and Tang[11] found that the rod block had a strong tendency to segregate near the surfaces, due to its less conformational entropy loss than the coil block. Again, Nowak and Vilgis[14] indicated that rod blocks would prefer to lie parallel to surface to gain entropy and to therefore lower their confinement energy. Similar phenomenon was observed by Huang et al[15]. Quite recently, the photovoltaic properties of RC BCPs were studied by Shah and Ganesan[16, 17]. They found that the lamellar morphology especially that oriented perpendicular to the confining surfaces exhibited more desirable characteristics than the nonlamellar morphologies like puck phase.
Experimentally, various studies have also been done to investigate the self-assembly of RC BCP films. Hierarchical structures of polystyrene-b-poly(γ-benzyl L-glutamate) (PS-b-PBLG) RC BCP films, including short ribbons, micron-sized lense shaped structures, a “twist grain boundary” (TGB)-like twisted smectic phase and so forth, have been reported by Ludwigs et al[18]. Park and Cho[19] reported recently the formation of strips in PS-b-polyisocyanate (PIC) RC BCP film where the strip width varied with the hydrophilicity of the substrate. Olsen et al.[20] found that the film thickness obviously affects the lamellae orientation in symmetric poly(alkoxyphenylenevinylene-b-isoprene) (PPV-b-PI) RC BCP thin films. Furthermore, the nanostructures of RC BCP films of poly(2, 5-diethylhexyloxy-1, 4-phenylenevinylene) (DEH-PPV) rods linking to different coil blocks, for their potential applications in the elaboration of photovoltaic devices, were studied by Heiser et al[21]. They obtained lamellar structure orienting either parallel or perpendicular to the surfaces by using a poly(n-butylacrylate) (PnBA) as coil block and found that switching to a glassy PS block would frustrate the ordering.
Relative to RC BCPs, the self-assembly of confined RCR triblock copolymers is far less explored both in experiments and theories. Contrast to RC BCPs, the arrangement of rod blocks in RCR triblock copolymers is strongly influenced by the behavior of the other rods in the polymer chain due to the connectivity of each block[22]. Thus, RCR triblock copolymers confined in thin films are expected to present unique self-assembly behavior. Recently, Kim’s group[23] observed liquid crystalline monolayers or bilayers of poly(n-hexyl isocyanate-b-2-vinylpyridine-b-n-hexyl isocyanate) (PHIC-b-P2VP-b-PHIC) RCR triblock copolymers on the coil-selective substrate. Park and his coworkers[24] reported the equilibrium structure of tetraaniline-b-poly(ethyleneglycol)-b-tetraaniline (TAN-b-PEG-b-TAN) RCR BCPs changed from multi-layered lamellae to disordered microdomains with the change of solvent. By using atomic force microscopy, Ibarboure et al.[25] found that PBLG-b-polydimethylsiloxane (PDMS)-b-PBLG RCR triblock copolymer films preferred to form lamellar structure in spite of the variation of the composition of BCPs. Monte Carlo (MC) simulation on the aggregation of RCR triblock copolymers between two impenetrable nonselective surfaces showed that RCR BCPs were likely to form perpendicular lamellae (L⊥) with increasing the rigidity of rod blocks[26]. A dissipative particle dynamics (DPD) simulation found that some special morphologies, such as wave lamellae parallel to the surfaces, orderly packed alternating cylinders and so on, can be formed by RCR triblock copolymers within a rod-selective slit[27].
In this paper, the self-assembly structure of sphere-forming RCR triblock copolymers within a planar slit is studied by DPD method. The paper is organized as follows: First we briefly describe the model and the simulation details. Then we investigate the effects of the slit property, the slit height, and the rod length on the equilibrium structures. Additionally, a phase diagram is given to describe the dependence of the self-assembled morphology on the height and property of the slit. The last section is devoted to conclusions.
SIMULATION DETAILS
DPD Algorithm
The DPD method, introduced by Hoogerbrugge and Koelman[28] in 1992, is a coarse-grained particle-based dynamic simulation technique that can use larger length and time scale. In this method, a group of molecules, or a group of segments of the polymer, are considered as a DPD particle. All the DPD particles interact with each other via pair-wise interactions that contain conservative force Fij(C), dissipative force Fij(D), and random force Fij(R). All DPD particles obey Newton's equation[29]
where mi, ri, vi, and fi denote the mass, position, velocity of the ith particle, and the acting force on it, respectively.
The total force on the ith particle, fi, is the sum of all pair-wise interactions as
The three pair-wise forces are given by:
where aij in Eq. (3) is the repulsive interaction between particles i and j, rij = ri - rj,
${{\boldsymbol{\hat r}}_{ij}} = {{\boldsymbol{r}}_{ij}}/{r_{ij}}$ , vij=vi-vj. Here θij is a symmetric random noise with zero mean and unit variance and is uncorrelated for different degrees of freedom and different time steps, σ is the amplitude of the thermal noise, and γ is a friction coefficient. The combined effect of the dissipative and random forces is that of a thermostat, leading to σ2 = 2γkBT. The weight function w(rij) provides the range of interactions for DPD particles with a commonly used choice: w(rij)=1-rij/rc for rij≤rc and w(rij)=0 for r > rc, where rc is the cutoff radius.In this work, the integration of the equations of motion is performed by using the modified velocity-Verlet algorithm[29] with λ = 0.65 and a time step Δt = 0.01. And we set σ = 3.67.
Models for Slit and RCR Copolymer Chain
Our simulations are focus on RxCyRx triblock copolymers confined between a planar slit composed of two impenetrable parallel walls separated by a distance of H. Each wall with a thickness of 1rc is constructed by four layers of DPD particles that are arranged in a face-centered cubic lattice. The centers of the wall DPD particles along the normal direction are placed at 1/8, 3/8, 5/8, and 7/8 of the wall. The high density of particles in the wall prevents other DPD particles from penetrating the wall, although the interactions between coarse-grained particles are soft. For simplicity, the DPD particles constructing the walls are motionless in the simulations.
For a polymer chain, a finitely extensible non-linear elastic (FENE) potential is added between the consecutive particles to bind the connected particles of the chain together[30]:
where the equilibrium bond length req = 0.8, the maximum bond length rmax = 1.3, and the elastic coefficient kF = 200.
To ensure the rigidity of the rod block, an additional bond-bending force between consecutive bonds is introduced as[31]
with kθ and θ0 = π are the bending modulus and the equilibrium angle between consecutive bonds, respectively. Here θ is the bond angle between two sequentially connected bonds. The rigidity of the rod block is represented by the value kθ. In this work, kθ is fixed at 100 with <θ> = 173°, therefore the block can be regarded as a rigid one.
The bond length distribution in R5C30R5 triblock copolymers has been investigated in bulk. A Gaussian distribution is observed and roughly the same for the rod and coil blocks, indicating that the distribution is determined by the FENE potential itself. The distribution peak and the average bond length are close to req = 0.8.
In our system, the number density of DPD particles is set to be 3.0. There are three types of DPD particles, including rod monomer (R) and coil monomer (C) in copolymer chains, and the slit surface (S). The repulsive interaction parameters aij between different types of DPD particles are chosen as:
Typically, the pair-wise repulsive interaction parameter between the same type of DPD particles is set as aii = 25 according to Ref. [29]. The interaction parameter between different particles i and j can be specified by the off-diagonal values. Obviously, monomers R and C repel each other with aRC = 32. And the interaction between copolymer and slit is represented by aRS and aCS.
In the present work, we mainly focus on R5C30R5 triblock copolymer whose volume fraction of rod block is fR = 0.25. In addition, an effective slit-copolymer interaction is introduced as Δa = aRS - aCS. Depending on the value of Δa, the slit is divided into three categories: (1) R-selective slit (Δa < 0, aRS = 5 - 25, aCS = 25); (2) Non-selective slit (Δa = 0, aRS = aCS = 25) and (3) C-selective slit (Δa > 0, aRS = 25, aCS = 5 - 25).
We have also investigated the effect of the rod length by keeping fR = 0.25 on the self-assembly behavior of RxCyRx within R-selective slits at H = 6, 10 and 12. The effect of system size (lateral size parallel to surface) on the assembly structure has been studied. It is found that the assembly structures are almost independent of the lateral size when it reaches 40 for 5≤x≤8. While Lx = Ly = 50 is sufficiently large for x= 9. Therefore, the effect of lateral size on the assembly structure is negligible in this work.
RESULTS AND DISCUSSION
At the beginning of the simulation, all polymers are randomly put into the system. Then we simulate the evolution of the system for a sufficient long time. During the simulation, we monitor the copolymer conformation and structure, and calculate the system energy at every 200τ. When the copolymer structure and the system energy only show small fluctuation with the simulation time, we assume the system is in equilibrium. It usually takes more than 80000τ for the formation of an equilibrium structure. And the time increases with the increase in the length of rod block. For x = 9, the dynamic process lasts for 100000τ. Then, we run additional 10000τ for statistics.
The phase behavior of BCPs is mainly controlled by two parameters, fR and aRC, which characterize the composition of the block copolymers and the degree of segregation, respectively. Here, we fix fR = 0.25 and aRC = 32. Such RCR triblock copolymers form sphere phase in bulk. Figure 1 shows the sphere structure assembled by R5C30R5 in bulk with aRC = 32. However, different structures are formed by R5C30R5 copolymers when they are confined within a slit. And the structure depends on the property (Δa) and height (H) of the slit. For each given H and Δa, we have simulated at least 5 independent runs. The same structure can be always observed.
Figure 1.
The sphere structure of R5C30R5 triblock copolymers in bulk Red and blue lines represent monomers R and C, respectively. Symbols are the same in the remaining figures. System size Lx = Ly = Lz = 30 and PBCs along all directions.
Effect of the Length of Rod Block
It is known that the length of the rod block has a significant effect on the phase behavior of block copolymers in solution[38]. Pryamitsyn and Ganesan[17] indicated that the effect of rod block was more evident for longer rod blocks and there was a critical rod length ≈ 8-9 inducing the disorder-order transition. Then we investigate the effect of the rod length on the self-assembly behavior of RxCyRx within R-selective slit. We fix fR = 0.25 by changing x = 6, 7, 8, 9, and the copolymers are expressed as R6C36R6, R7C42R7, R8C48R8, and R9C54R9. Three heights of slit with H = 6, 10 and 12 are considered.
When RxCyRx triblock copolymers are confined in a narrow slit (H = 6), W structures are always formed irrespective of the rod length. However, we find that the degree of rod alignment in W increases by changing x from 5 to 9. Seen from Fig. 8, short rods appear to be randomly oriented. Whereas long rods, such as x = 7, 8 and 9, are arranged in an ordered orientation.
 Figure 8.
Top view of the equilibrium structure of (a) R5C30R5 and (b) R9C54R9 triblock copolymers formed at H = 6 in R-selective slit (Δa = -20)
Figure 8.
Top view of the equilibrium structure of (a) R5C30R5 and (b) R9C54R9 triblock copolymers formed at H = 6 in R-selective slit (Δa = -20)
To quantify the alignment of rod blocks within W layer, a rod-rod orientational order parameter is calculated for the rod blocks. We define the rod-rod orientational order parameter as[39]
Here θij is the included angle between two rods, rij is the distance between mass centers of rod i and rod j. A sketch for θij, and rij is presented in Fig. 9. The delta function is defined as
In addition, we introduce a condition function
$f{\rm{(}}{\vec r_i}{\rm{, }}{\vec r_j}{\rm{)}}$ in calculating s(r). For the wetting layers formed in our system, we have$f{\rm{(}}{\vec r_i}{\rm{, }}{\vec r_j}{\rm{)}}$ = 1 for both rods being in the same layer and$f{\rm{(}}{\vec r_i}{\rm{, }}{\vec r_j}{\rm{)}}$ = 0 otherwise. Thus, s(r) is calculated in each layer and further averaged over two layers near upper and lower surfaces.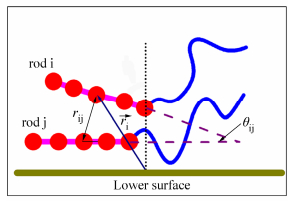 Figure 9.
A sketch of qij and rij near surface (qij and rij are the included angle between two rods and the distance between mass centers of rod i and rod j, respectively.)
Figure 9.
A sketch of qij and rij near surface (qij and rij are the included angle between two rods and the distance between mass centers of rod i and rod j, respectively.)
The rod-rod orientational order parameter s(r) represents the space correlation between rod blocks. We have s = 0 for a completely random and isotropic sample and s = 1 for a perfectly aligned sample. The dependence of orientational order parameter s(r) on rod-rod distance r in W for RxCyRx triblock copolymers is presented in Fig. 10(a). For x < 7, s is small and decays fast with distance, showing that the rods are roughly randomly oriented in long length scale. However, for x > 7, s is large and decays slowly with distance, indicating an ordered arrangement for rod blocks. It was pointed out that, for a typical liquid crystal sample, s is on the order of 0.3 to 0.8[40]. Our results show that there is long-range orientation order for x > 7 and only short-range order for x < 7. The existence of short-range orientation order for rod length x < 7 is obviously different from that of copolymers in solution[39]. So we suppose the slit can help the rod blocks to establish short-range orientation order. The mean orientational order parameter <s> for different rod lengths x is presented in Fig. 10(b). Here <s> is averaged over all rod-rod pairs in Ws. We find that <s> increases obviously from x = 7. So we conclude the critical rod length for the disorder-order transition of rod orientation in the W structure is x* = 7, which is smaller than that of polymer solution[17]. Our results show that the confinement of slit plays an important role in the disorder-order transition of rod orientation in RCR copolymers.
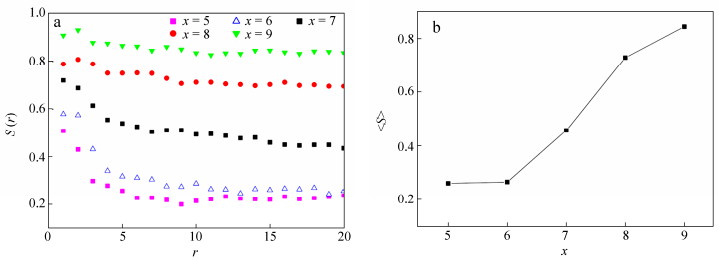 Figure 10.
(a) The orientational order parameter s(r) and (b) mean orientational order parameter <s> of rod blocks in W for RxCyRx triblock copolymers within R-selective slit with Δa = -20 and H = 6
Figure 10.
(a) The orientational order parameter s(r) and (b) mean orientational order parameter <s> of rod blocks in W for RxCyRx triblock copolymers within R-selective slit with Δa = -20 and H = 6
When the slit height increases to H = 10, the rod blocks in W form different aggregations with increasing the rod length. For x = 5-7, they form island-like structure. And arrowhead-shaped (AH) morphology is formed at x = 8 and 9. The structures formed at x = 6 and 8 are shown in Fig. 11(a).
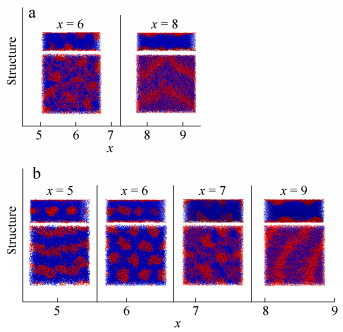 Figure 11.
The assembled structures of RxCyRx triblock copolymers in R-selective slits (Δa = -20) with various rod length x at H = (a) 10 and (b) 12
Figure 11.
The assembled structures of RxCyRx triblock copolymers in R-selective slits (Δa = -20) with various rod length x at H = (a) 10 and (b) 12
When H increases to 12, the assembled structure changes obviously with the rod length x, as shown in Fig. 11(b). As we can see, C||, 1 and C⊥ between wetting layers are formed by R5C30R5 and R6C36R6 copolymers, respectively. Island-like structure is formed at x = 7. When the rod length increases to 8 and 9, parallel half-cylinders (C||, 1/2) near surfaces are assembled by rod blocks. It was supposed that a small degree of orientational freedom may stabilize the semi-cylinder structure[41]. Our results show that the rod length strongly influences the assembled structure.
R-selective Slit
In this section, R5C30R5 triblock copolymers are confined within R-selective slits with Δa = -20. The effect of the slit height H on the self-assembly of R5C30R5 copolymers is investigated by varying H from 3 to 24. And some typical structures are presented in Fig. 2.
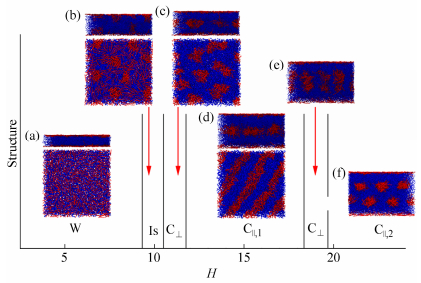 Figure 2.
Phase diagram of R5C30R5 triblock copolymers in R-selective slits with Δa = -20: H = 6 (a), 10 (b), 11 (c), 16 (d), 19 (e) and 20 (f) For (a)-(b), side view (top) and top view (bottom) are presented; For (c)-(d), side view (top) and cross-section view of the system at the center (bottom) are presented; Only side view is presented for (e) and (f).
Figure 2.
Phase diagram of R5C30R5 triblock copolymers in R-selective slits with Δa = -20: H = 6 (a), 10 (b), 11 (c), 16 (d), 19 (e) and 20 (f) For (a)-(b), side view (top) and top view (bottom) are presented; For (c)-(d), side view (top) and cross-section view of the system at the center (bottom) are presented; Only side view is presented for (e) and (f).
In narrow slits with H≤9, wetting layer (W) of rod blocks is always formed near slit surface, as shown in Fig. 2(a). In fact, W is always observed in all R-selective slits. The appearance of W can be attributed to the energy favor of rod blocks. When the slit height increases to be H = 10, the rod blocks in W begin to aggregate and penetrate into the inner coil blocks to form island-like structure (Is), see Fig. 2(b). With a further increase of H, the aggregation disappears, and new structures between two wetting layers are formed. For H = 11, perpendicular cylinders (C⊥) are formed at the center of slit (Fig. 2c). And one layer of parallel cylinders (C||, 1) are assembled by rod blocks in the region of H = 12-18, as shown in Fig. 2(d). However, when H increases to be 19, C⊥ is formed again. Afterwards, two-layer parallel cylinders (C||, 2) are formed at H = 20-24. Figure 2(f) shows the equilibrium morphology assembled at H = 20.
In order to get a further insight into the self-assembly behavior of R5C30R5 triblock copolymers within R-selective slit, we have calculated the mean square end-to-end distance <R2>, the mean square radius of gyration <Rg2> as well as their horizontal and vertical components. The dependence of <R2> and <Rg2>, as well as their horizontal and vertical components on the slit height H is plotted in Fig. 3.
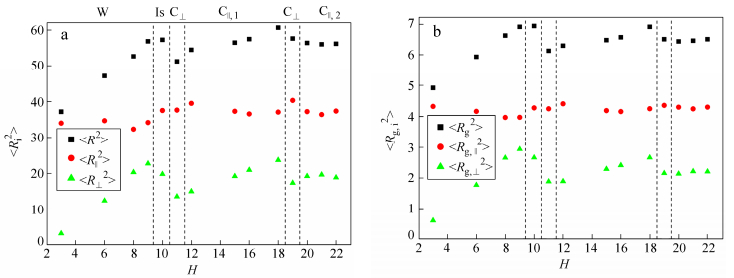 Figure 3.
(a) The mean square end-to-end distance <R2>, and its horizontal <R||2> and vertical <R⊥2> component; (b) The mean square radius of gyration <Rg2>, and its horizontal <Rg, ||2> and vertical<Rg, ⊥2> component of R5C30R5 triblock copolymers as a function of the slit height H (Δa = -20)
Figure 3.
(a) The mean square end-to-end distance <R2>, and its horizontal <R||2> and vertical <R⊥2> component; (b) The mean square radius of gyration <Rg2>, and its horizontal <Rg, ||2> and vertical<Rg, ⊥2> component of R5C30R5 triblock copolymers as a function of the slit height H (Δa = -20)
As shown in Fig. 3, the transition of structures is always accompanied by an obvious change in the conformational properties of R5C30R5 triblock copolymers. In narrow slits where the system forms W, the polymer chain becomes increasingly elongated in the vertical direction (i.e., <R⊥2> and <Rg, ⊥2> increase) as H increases. This behavior is associated with a release of the steric constraint imposed by the confining surfaces. For H = 10, <R2> and <Rg2> are almost unchanged, but <R||2> and <Rg, ||2> increase with a sudden decrease of <R⊥2> and <Rg, ⊥2>, indicating the orientation change of copolymers. At H = 11, <R2> and <Rg2> as well as their vertical components <R⊥2> and <Rg, ⊥2> all decreased steeply, corresponding to the formation of C⊥. With a further increase of H, the transformation into parallel cylinders is accompanied by a dramatic elongation of polymer chain in the vertical direction, as evidenced by the increase of <R⊥2> and <Rg, ⊥2> with H within C||, 1 phase. As the slit height is increased, the structural frustration of R5C30R5 in C||, 1 is relieved by either formation of an additional parallel cylinders (C||, 2) or a reorientation of cylinders to form C⊥. Similar to Pavel and John[32], who found that the structural frustration of lamellae- and cylinder-forming BCPs in planar slits was alleviated by increasing the layers of parallel structure or forming perpendicular structure. Therefore, we get a conclusion that the self-assembly of RCR triblock copolymer are strongly dependent on the slit height which influences the conformation and orientation of polymer chains.
Surprisingly, the formation process of C||, 1 at H = 18 is different from others formed at H < 18. More than ten independent samples have been simulated and similar process is observed at H = 18. R5C30R5 copolymers firstly form C⊥, these cylinders then try to connect with each other in the horizontal direction, and C||, 1 are finally formed. Some typical snapshots captured at various times are shown in Fig. 4(a). However, C||, 1 is evolved from defective cylinders for H < 18. Figure 4(b) presents some typical structures formed at different times for H = 15. We suppose that the different formation process of C||, 1 at H = 18 may be due to the slit height close to 19 where C⊥ is formed, leading to the competition between the formation of C||, 1 and C⊥, and C||, 1 is formed ultimately.
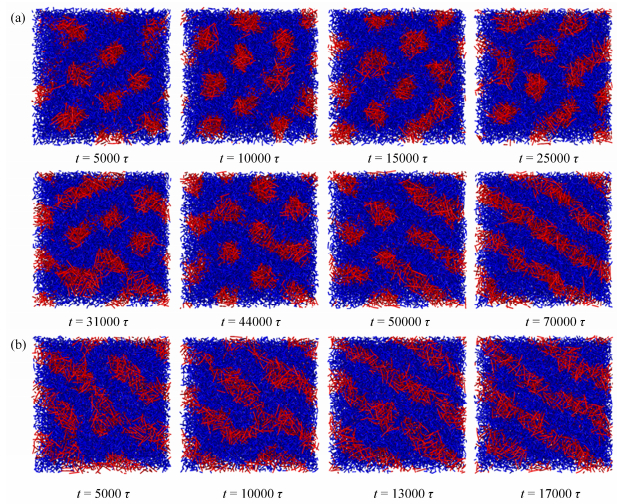 Figure 4.
Simulation snapshots showing the evolution of parallel cylinders within R-selective slit (Δa = -20) for H = 18 (a) and 15 (b)
Figure 4.
Simulation snapshots showing the evolution of parallel cylinders within R-selective slit (Δa = -20) for H = 18 (a) and 15 (b)
C-selective Slits
Meanwhile, we have also investigated the self-assembly behavior of R5C30R5 copolymers within C-selective slits (Δa = 20). The equilibrium structures formed at different slit heights are presented in Fig. 5. At H = 3-4, unlike the wetting layers formed in R-selective slit, C⊥ is formed in strong C-selective slit. When H increases to be 5-10, C||, 1 are formed, as shown in Fig. 5(a). With a further increase of H, alternating C⊥ and C||, 2 are observed, see Figs. 5(b) and 5(c). We find that the layer number of C|| increases with H. However, C⊥ is not formed when C||, 1 transforms into C||, 2. Rather, the cross-cylindrical structure (C×) in which the orientation of cylinders in different layers is different but both parallel to surfaces are formed for H = 20. In Fig. 5(d), the side view of C× and the cross-section view of top and bottom layer are presented respectively. With further increase in H, the C||, 3 structure is formed. Figure 5(e) shows the side view of the equilibrium morphology assembled at H = 22. Obviously, parallel cylinders are in an approximately hexagonal arrangement along the slit height direction.
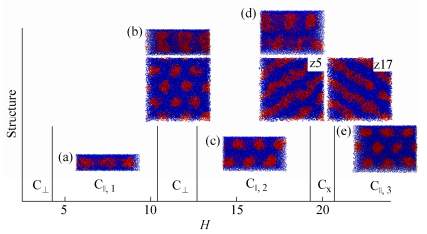 Figure 5.
Phase diagram of R5C30R5 copolymers in strong C-selective slits with Δa = 20 for H = 8 (a), 12 (b), 16 (c), 20 (d) and 22 (e) Side views are presented in (a), (c) and (e); side view (top) and cross-section view (bottom) of the system are presented in (b) and (d).
Figure 5.
Phase diagram of R5C30R5 copolymers in strong C-selective slits with Δa = 20 for H = 8 (a), 12 (b), 16 (c), 20 (d) and 22 (e) Side views are presented in (a), (c) and (e); side view (top) and cross-section view (bottom) of the system are presented in (b) and (d).
The self-assembly behavior of confined flexible A3B12A3 triblock copolymers which formed spheres in bulk has been investigated by Sevink and Zvelindovsky[33]. They found that alternating spheres and C⊥ were formed by increasing the slit height. In their study, the slit is preferential for the middle block as well. Whereas C||, v is formed by RCR triblock copolymers in our system. It is known that sphere is more sensitive to frustration in one direction than cylinder, due to its 3D nature and reduced deformability. And a long chain will experience a larger entropic penalty because of the presence of the rod blocks in both ends. By increasing the interaction between copolymer and slit surface, C||, v is formed in their system as well. However, C× was not observed by Sevink and Zvelindovsky. Shi et al.[34] observed C× occurring just before the increase of the layer number of C||, indicating that C× is the most frustrated structure. However, no C⊥ structure was assembled by flexible A2B10 copolymers in their simulations. The formation of C× structure in our simulation may also due to the volume fraction of rod block fR = 0.25 very close to the order-to-order (OOT) transition from sphere to cylinder in bulk.
Horvat et al.[35] have studied the evolution of local morphology and terrace formation in block copolymer films with incommensurate thickness. They found that a vertical orientation of cylinders evolved into a parallel one via deformed necks as intermediate structure, and the formation of the second layer of parallel cylinders was achieved by interconnecting the deformed necks. The interconnection can start either parallel to the cylinders in the bottom layer or under an angle. In our case, C× structure is formed through the interconnection under an angle.
We have also calculated the dependence of <R2> and <Rg2> as well as their horizontal and vertical components on H. As shown in Fig. 6, the polymer chain is increasingly elongated in the perpendicular direction with an increase in H, as evidenced by the increase of <R⊥2> and <Rg, ⊥2>. However, <R||2> and <Rg, ||2> increase or decrease suddenly corresponding to the formation of C⊥ and C×. It demonstrates that the transformation into a particular phase, such as C⊥ and C×, is accompanied by a change in the conformation of polymer chains. Comparing Fig. 6 with Fig. 3, we find the slit property also influences the conformation of polymer chains. For instance, when the assembled structure changes from C⊥ to C||, 1, <R2> and <Rg2> both increase with H in R-selective slits, indicating the polymer chain stretches; while in C-selective slits, the polymer chain is shrunk, as evidenced by the decrease of <R2> and <Rg2> with increasing H.
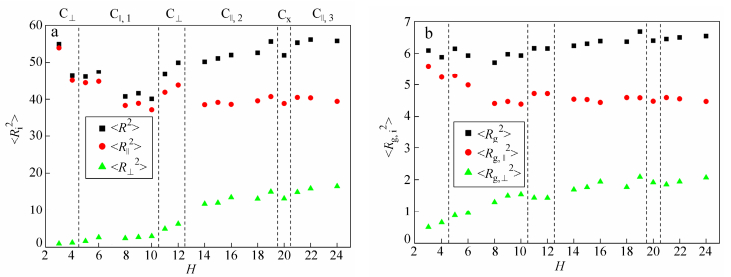 Figure 6.
(a) The mean square end-to-end distance <R2>, and its horizontal <R||2> and vertical <R⊥2> component; (b) The mean square radius of gyration <Rg2>, and its horizontal <Rg, ||2> and vertical<Rg, ⊥2> component, as a function of the slit height H for R5C30R5 copolymers within C-selective slits (Δa = 20)
Figure 6.
(a) The mean square end-to-end distance <R2>, and its horizontal <R||2> and vertical <R⊥2> component; (b) The mean square radius of gyration <Rg2>, and its horizontal <Rg, ||2> and vertical<Rg, ⊥2> component, as a function of the slit height H for R5C30R5 copolymers within C-selective slits (Δa = 20)
Phase Diagram
The dependence of the self-assembled structures on the slit height H and the slit property (Δa) are then summarized in a phase diagram shown in Fig. 7. We find that the structures assembled by R5C30R5 triblock copolymer within non-selective slits (Δa = 0) are analogous to that within R-selective slits except for H = 3. C⊥ is formed within a non-selective slit at H = 3. Huinink et al.[36] and Wang et al.[37] have found that the neutral wall has weak preference for the short block because of the entropic effect. Meanwhile, the rod blocks would prefer to lie parallel to the surfaces[12]. Consequently, the non-selective slit can be considered as a weak R-selective one. And the different structure formed at H = 3 can be contributed to the short-range effect of slit surface. For the weak C-selective slits (0 < Δa≤10), C⊥ is always formed regardless of the change of H. So we think that the surface can be regarded as completely neutral in the vicinity of Δa = 10. For strong C-selective slits (Δa ≥ 15), alternating C⊥(C×) and C//, v are observed with H increasing from 3 to 24. Ultimately, we conclude that the self-assembled structure of confined system is not only dependent on the property but also on the height of the slit.
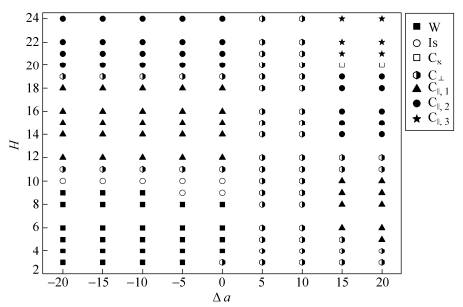 Figure 7.
Phase diagram of structures assembled by R5C30R5 copolymers within a planar slit with respect to the slit height (H) and the slit property (Δa)
Figure 7.
Phase diagram of structures assembled by R5C30R5 copolymers within a planar slit with respect to the slit height (H) and the slit property (Δa)
CONCLUSIONS
In this paper, dissipative particle dynamics simulations are performed to systematically explore the self-assembly of sphere-forming R5C30R5 triblock copolymers within a slit. A phase diagram of the self-assembled structures with respect to the slit height (H) and the slit property (Δa) is presented. A sequence of novel structures which are not seen in bulk are formed by varying Δa and H. There are five main structures in the phase diagram, including wetting layers (W), island-like (Is) structure, parallel cylinders (C||, v), perpendicular cylinders (C⊥) and cross-cylindrical structures (C×). C⊥ structure is always formed before the number of C|| layers is increased.
The rod length is found to have an important impact on the rod alignment, and the orientational order parameter grows significantly as the rod length increases. Some special structures, such as parallel half-cylinders (C||, 1/2) and arrowhead-shaped (AH) morphology, are observed when the rod length increases to a certain extent. The results in this study may provide a guide to understand the interplay among the chain conformation, the confinement effect, the surface field and the rod length in thin films of RCR triblock copolymers.
-
-
[1]
Matsen, M.W. and Schick, M., Phys. Rev. Lett., 1994, 72: 2660
-
[2]
Tuzar, Z. and Kratochvil, P., "Micelles of block- and graft-copolymers in solutions", Plenum Press, New York, 1993
-
[3]
Chen, J.Z., Zhang, C.X., Sun, Z.Y. and An, L.J., J. Chem. Phys., 2007, 127: 024105
-
[4]
Matsen, M.W. and Thompson, R.B., J. Chem. Phys., 1999, 111: 7139
-
[5]
Wang, Q., Nealey, P.F. and de Pablo, J.J., Macromolecules, 2003, 36: 1731
-
[6]
Chai, A.H. and Zhang, L.X., Chinese J. Polym. Sci., 2011, 29(6): 684
-
[7]
Yin, Y., Sun, P., Jiang, R. and Li, B.H., J. Chem. Phys., 2006, 124: 184708
-
[8]
Chen, J.T., Thomas, E.L., Ober, C.K. and Mao, G., Science, 1996, 273: 343
-
[9]
Shi, L.Y., Pan, Y., Zhang, Q.K., Zhou, Y., Fan, X.H. and Shen, Z.H., Chinese J. Polym. Sci., 2014, 32(11): 1524
-
[10]
Pereira, G.G. and Williams, D.R.M., Macromolecules, 2000, 33: 3166
-
[11]
Yang, G., Tang, P., Yang, Y.L. and Wang, Q., J. Phys. Chem. B, 2010, 114: 14897
-
[12]
Wang, Q., Soft Matter, 2011, 7: 3711
-
[13]
Cheng, L.S. and Cao, D.P., J. Chem. Phys., 2008, 128: 074902
-
[14]
Nowak, C. and Vilgis, T.A., J. Chem. Phys., 2006, 124: 234909
-
[15]
Huang, J.H., Ma, Z.X. and Luo, M.B., Langmuir, 2014, 30: 6267
-
[16]
Shah, M. and Ganesan, V., Macromolecules, 2010, 43: 543
-
[17]
Pryamitsyn, V. and Ganesan, V., J. Chem. Phys., 2004, 120: 5824
-
[18]
Ludwigs, S., Krausch, G., Antonietti, M. and Schlaad, H., Macromolecules, 2005, 38: 7532
-
[19]
Park, J.W. and Cho, Y.H., Langmuir, 2006, 22: 10898
-
[20]
Olsen, B.D., Li, X., Wang, J. and Segalman, R.A., Macromolecules, 2007, 40: 3287
-
[21]
Heiser, T., Adamopoulos, G., Brinkmann, M. and Hadziioannou, G., Thin Solid Films, 2006, 511: 219
-
[22]
Xia, Y.D., Chen, J.Z., Shi, T.F. and An, L.J., Chinese J. Polym. Sci., 2013, 31(9): 1242
-
[23]
Kim, J.H., Rahman, M.S., Lee, J.S. and Park, J.W., Macromolecules, 2008, 41: 3181
-
[24]
Kim, H., Kim, T.G. and Park, J.W., Macromol. Res., 2013, 21: 815
-
[25]
Ibarboure, E. and Rodríguez-Hernández, J., Eur. Polym. J., 2010, 46: 891
-
[26]
Cui, J., Zhu, J.T., Ma, Z.W. and Jiang, W., Chem. Phys., 2006, 321: 1
-
[27]
Ma, Z.X., Huang, J.H. and Luo, M.B., Soft Matter, 2015, 11: 4932
-
[28]
Hoogerbrugge, P.J. and Koelman, J.M.V.A., Europhys. Lett., 1992, 19: 155
-
[29]
Groot, R.D. and Warren, P.B., J. Chem. Phys., 1997, 107: 4423
-
[30]
Kremer, K. and Grest, G.S., J. Chem. Phys.1990, 92: 5057
-
[31]
AlSunaidi, A., den Otter, W.K. and Clarke, J.H.R., Phil. Trans. R. Soc. London. A, 2004, 362: 1773
-
[32]
Petrus, P., Lisal, M. and Brennan, J.K., Langmuir, 2010, 26: 14680
-
[33]
Sevink, G.J.A. and Zvelindovsky, A.V., Macromolecules, 2009, 42: 8500
-
[34]
Yu, B., Li, B.H., Jin, Q.H., Ding, D.T. and Shi, A.C., Soft Matter, 2011, 7: 10227
-
[35]
Horvat, A., Knoll, A., Krausch, G. and Tsarkova, L., Macromolecules, 2007, 40: 6930
-
[36]
Huinink, H.P., van Dijk, H.P.M.A., Brokken-Zijp, J.C.M. and Sevink, G.J.A., Macromolecules, 2001, 34: 5325
-
[37]
Wang, Q., Nealey, P.F. and de Pablo, J.J., Macromolecules, 2001, 34: 3458
-
[38]
Tu, Y.F., Wan, X.H., Zhang, H., L., Fan, X.H., Lü, D.N., Chen, X.F. and Zhou, Q.F., Chinese J. Polym. Sci., 2003, 21(5): 569
-
[39]
Chou, S.H., Tsao, H.K. and Sheng, Y.J., J. Chem. Phys., 2011, 134: 034904
-
[40]
Horsch, M.A., Zhang, Z. and Glotzer, S.C., Nano Lett., 2006, 6: 2406
-
[41]
Horsch, M.A., Zhang, Z. and Glotzer, S.C., Soft Matter, 2010, 6: 945
-
[1]
-

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1907
- HTML全文浏览量: 53
 下载:
下载:
