-
[1]
Dinh, K. N.; Liang, Q.; Du, C. F.; Zhao, J.; Tok, A. I. Y.; Mao, H.; Yan, Q. Nanostructured metallic transition metal carbides, nitrides, phosphides, and borides for energy storage and conversion. Nano. Today 2019, 25, 99−121. doi: 10.1016/j.nantod.2019.02.008
-
[2]
Gupta, A.; Sakthivel, T.; Seal, S. Recent development in 2D materials beyond graphene. Prog. Mater. Sci. 2015, 73, 44−126. doi: 10.1016/j.pmatsci.2015.02.002
-
[3]
Mortazavi, B.; Dianat, A.; Cuniberti, G.; Rabczuk, T. Application of silicene, germanene and stanene for Na or Li ion storage: a theoretical investigation. Electrochim. Acta 2016, 213, 865−870. doi: 10.1016/j.electacta.2016.08.027
-
[4]
Oughaddou, H.; Enriquez, H.; Tchalala, M. R.; Yildirim, H.; Mayne, A. J.; Bendounan, A.; Dujardin, G.; Ait Ali, M.; Kara, A. Silicene, a promising new 2D material. Prog. Surf. Sci. 2015, 90, 46−83. doi: 10.1016/j.progsurf.2014.12.003
-
[5]
Zhuang, J.; Xu, X.; Feng, H.; Li, Z.; Wang, X.; Du, Y. Honeycomb silicon: a review of silicene. Sci. Bull. 2015, 60, 1551−1562. doi: 10.1007/s11434-015-0880-2
-
[6]
Ali, M.; Ni, Z.; Cottenier, S.; Liu, Y.; Pi, X.; Yang, D. Formation, structures and electronic properties of silicene oxides on Ag(111). J. Mater. Sci. Technol. 2016, 33, 751−757.
-
[7]
Zhao, J.; Liu, H.; Yu, Z.; Quhe, R.; Zhou, S.; Wang, Y.; Liu, C. C.; Zhong, H.; Han, N.; Lu, J.; Yao, Y.; Wu, K. Rise of silicene: a competitive 2D material. Prog. Mater. Sci. 2016, 83, 24−151. doi: 10.1016/j.pmatsci.2016.04.001
-
[8]
Molle, A.; Grazianetti, C.; Tao, L.; Taneja, D.; Alam, M. H.; Akinwande, D. Silicene, silicene derivatives, and their device applications. Chem. Soc. Rev. 2018, 47, 6370−6387. doi: 10.1039/C8CS00338F
-
[9]
Lee, J. H.; Lee, E. K.; Joo, W. J.; Jang, Y.; Kim, B. S.; Lim, J. Y.; Choi, S. H.; Ahn, S. J.; Ahn, J. R.; Park, M. H. Wafer-scale growth of single-crystal monolayer graphene on reusable hydrogen-terminated germanium. Science 2014, 344, 286−289. doi: 10.1126/science.1252268
-
[10]
Ni, Z.; Minamitani, E.; Ando, Y.; Watanabe, S. The electronic structure of quasi-free-standing germanene on monolayer MX (M = Ga, In; X = S, Se, Te). Phys. Chem. Chem. Phys. 2015, 17, 19039−19044. doi: 10.1039/C5CP02428E
-
[11]
Sa, B.; Li, Y. L.; Qi, J.; Ahuja, R.; Sun, Z. Strain engineering for phosphorene: the potential application as a photocatalyst. J. Phys. Chem. C 2014, 118, 26560−26568. doi: 10.1021/jp508618t
-
[12]
Li, L.; Yu, Y.; Ye, G. J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X. H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372−377. doi: 10.1038/nnano.2014.35
-
[13]
Wang, Q. H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J. N.; Strano, M. S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699−712. doi: 10.1038/nnano.2012.193
-
[14]
Peng, Q.; Wang, Z.; Sa, B.; Wu, B.; Sun, Z. Electronic structures and enhanced optical properties of blue phosphorene/transition metal dichalcogenides van der Waals heterostructures. Sci. Rep. 2016, 6, 31994−10. doi: 10.1038/srep31994
-
[15]
Guo, Z.; Zhou, J.; Zhu, L.; Sun, Z. MXene: a promising photocatalyst for water splitting. J. Mater. Chem. A 2016, 4, 11446−11452. doi: 10.1039/C6TA04414J
-
[16]
Naguib, M.; Mochalin, V. N.; Barsoum, M. W.; Gogotsi, Y. MXenes: a new family of two-dimensional materials. Adv. Mater. 2014, 26, 982−982. doi: 10.1002/adma.201470041
-
[17]
Rehman, G.; Khan, S. A.; Amin, B.; Ahmad, I.; Gan, L. Y.; Maqbool, M. Intriguing electronic structures and optical properties of two-dimensional van der Waals heterostructures of Zr2CT2 (T = O, F) with MoSe2 and WSe2. J. Mater. Chem. C 2018, 6, 2830−2839. doi: 10.1039/C7TC05963A
-
[18]
Sone, J.; Yamagami, T.; Aoki, Y.; Nakatsuji, K.; Hirayama, H. Epitaxial growth of silicene on ultra-thin Ag(111) films. New J. Phys. 2012, 16, 095004−15.
-
[19]
Meng, L.; Wang, Y.; Zhang, L.; Du, S.; Wu, R.; Li, L.; Zhang, Y.; Li, G.; Zhou, H.; Hofer, W. Buckled silicene formation on Ir(111). Nano. Lett. 2013, 13, 685−690. doi: 10.1021/nl304347w
-
[20]
Cherukara, M. J.; Narayanan, B.; Chan, H.; Skrs, S. Silicene growth through island migration and coalescence. Nanoscale 2017, 9, 10.1039. C1037NR03153J, 10186−10192.
-
[21]
Aizawa, T.; Suehara, S.; Otani, S. Silicene on zirconium carbide (111). J. Phys. Chem. C 2014, 118, 23049−23057. doi: 10.1021/jp505602c
-
[22]
Gill, T. G.; Fleurence, A.; Warner, B.; Prüser, H.; Friedlein, R.; Sadowski, J. T.; Hirjibehedin, C. F.; Yamadatakamura, Y. Metallic atomically-thin layered silicon epitaxially grown on silicene/ZrB2. 2D Mater. 2017, 4, 021015−7. doi: 10.1088/2053-1583/aa5a80
-
[23]
Feng, J. W.; Liu, Y. J.; Wang, H. X.; Zhao, J. X.; Cai, Q. H.; Wang, X. Z. Gas adsorption on silicene: a theoretical study. Comp. Mater. Sci. 2014, 87, 218−226. doi: 10.1016/j.commatsci.2014.02.025
-
[24]
Wei, H.; Nan, X.; Xiaojun, W.; Zhenyu, L.; Jinlong, Y. Silicene as a highly sensitive molecule sensor for NH3, NO and NO2. Phys. Chem. Chem. Phys. 2014, 16, 6957−6962. doi: 10.1039/c3cp55250k
-
[25]
Chandiramouli, R.; Srivastava, A.; Nagarajan, V. NO adsorption studies on silicene nanosheet: DFT investigation. Appl. Surf. Sci. 2015, 351, 662−672. doi: 10.1016/j.apsusc.2015.05.166
-
[26]
Ni, Z.; Zhong, H.; Jiang, X.; Quhe, R.; Luo, G.; Wang, Y.; Ye, M.; Yang, J.; Shi, J.; Lu, J. Tunable band gap and doping type in silicene by surface adsorption: towards tunneling transistors. Nanoscale 2014, 6, 7609−7618. doi: 10.1039/C4NR00028E
-
[27]
Tao, L.; Cinquanta, E.; Chiappe, D.; Grazianetti, C.; Fanciulli, M.; Dubey, M.; Molle, A.; Akinwande, D. Silicene field-effect transistors operating at room temperature. Nat. Nanotechnol. 2015, 10, 227−231. doi: 10.1038/nnano.2014.325
-
[28]
Le Lay, G. Silicene transistors. Nat. Nanotechnol. 2015, 10, 202−203. doi: 10.1038/nnano.2015.10
-
[29]
Yang, K.; Cahangirov, S.; Cantarero, A.; Rubio, A.; D'Agosta, R. Thermoelectric properties of atomically thin silicene and germanene nanostructures. Phys. Rev. B 2014, 89, 125403−13. doi: 10.1103/PhysRevB.89.125403
-
[30]
Wierzbicki, M.; Barnaś, J.; Swirkowicz, R. Zigzag nanoribbons of two-dimensional silicene-like crystals: magnetic, topological and thermoelectric properties. J. Phys. Condens. Mat. 2015, 27, 485301−14. doi: 10.1088/0953-8984/27/48/485301
-
[31]
Jose, D.; Datta, A. Structures and electronic properties of silicene clusters: a promising material for FET and hydrogen storage. Phys. Chem. Chem. Phys. 2011, 13, 7304−7311. doi: 10.1039/c0cp02580a
-
[32]
Jinglan, Q.; Huixia, F.; Yang, X.; Oreshkin, A. I.; Tingna, S.; Hui, L.; Sheng, M.; Lan, C.; Kehui, W. Ordered and reversible hydrogenation of silicene. Phys. Rev. Lett. 2015, 114, 126101−126105. doi: 10.1103/PhysRevLett.114.126101
-
[33]
Wang, Y.; Rui, Z.; Gao, H.; Jing, Z.; Xu, B.; Qiang, S.; Yu, J. Metal adatoms-decorated silicene as hydrogen storage media. Int. J. Hydrogen Energ. 2014, 39, 14027−14032. doi: 10.1016/j.ijhydene.2014.06.164
-
[34]
Xin, T.; Cabrera, C. R.; Chen, Z. Metallic BSi3 silicene: a promising high capacity anode material for lithium-ion batteries. J. Phys. Chem. C 2014, 118, 25836−25843. doi: 10.1021/jp503597n
-
[35]
Hwang, Y.; Yun, K. H.; Chung, Y. C. Carbon-free and two-dimensional cathode structure based on silicene for lithium-oxygen batteries: a first-principles calculation. J. Power Sources 2015, 275, 32−37. doi: 10.1016/j.jpowsour.2014.11.016
-
[36]
Rugeramigabo, E. P.; Nagao, T.; Pfnür, H. Experimental investigation of two-dimensional plasmons in a DySi2 monolayer on Si(111). Phys. Rev. B 2008, 78, 155402−6. doi: 10.1103/PhysRevB.78.155402
-
[37]
Yang, L. M.; Bačić, V.; Popov, I. A.; Boldyrev, A. I.; Heine, T.; Frauenheim, T.; Ganz, E. Two-dimensional Cu2Si monolayer with planar hexacoordinate copper and silicon bonding. J. Am. Chem. Soc. 2015, 137, 2757−2762. doi: 10.1021/ja513209c
-
[38]
Yang, L. M.; Popov, I. A.; Boldyrev, A. I.; Heine, T.; Frauenheim, T.; Ganz, E. Post-anti-van't Hoff-Le Bel motif in atomically thin germanium-copper alloy film. Phys. Chem. Chem. Phys. 2015, 17, 17545−17551. doi: 10.1039/C5CP02827B
-
[39]
Yang, L. M.; Popov, I. A.; Frauenheim, T.; Boldyrev, A. I.; Heine, T.; Bačić, V.; Ganz, E. Revealing unusual chemical bonding in planar hyper-coordinate Ni2Ge and quasi-planar Ni2Si two-dimensional crystals. Phys. Chem. Chem. Phys. 2015, 17, 26043−26048. doi: 10.1039/C5CP04893A
-
[40]
Feng, B.; Fu, B.; Kasamatsu, S.; Ito, S.; Cheng, P.; Liu, C. C.; Feng, Y.; Wu, S.; Mahatha, S. K.; Sheverdyaeva, P.; Moras, P.; Arita, M.; Sugino, O.; Chiang, T. C.; Shimada, K.; Miyamoto, K.; Okuda, T.; Wu, K.; Chen, L.; Yao, Y.; Matsuda, I. Experimental realization of two-dimensional Dirac nodal line fermions in monolayer Cu2Si. Nat. Commun. 2017, 8, 1007−6. doi: 10.1038/s41467-017-01108-z
-
[41]
Sun, Y.; Zhuo, Z.; Wu, X.; Yang, J. Room-temperature ferromagnetism in two-dimensional Fe2Si nanosheet with enhanced spin-polarization ratio. Nano. Lett. 2017, 17, 2771−2777. doi: 10.1021/acs.nanolett.6b04884
-
[42]
Wang, Y.; Qiao, M.; Li, Y.; Chen, Z. A two-dimensional CaSi monolayer with quasi-planar pentacoordinate silicon. Nanoscale Horiz. 2018, 3, 327−334. doi: 10.1039/C7NH00091J
-
[43]
Exner, K.; Schleyer, P. V. R. Planar hexacoordinate carbon: a viable possibility. Science 2000, 290, 1937−1940. doi: 10.1126/science.290.5498.1937
-
[44]
Wen, C.; Xie, Q.; Xiong, R.; Sa, B.; Miao, N.; Zhou, J.; Wu, B.; Sun, Z. Computational mining of the pressure effect on thermodynamic and thermoelectric properties of cubic Ca2Si. EPL 2018, 123, 67003−6. doi: 10.1209/0295-5075/123/67003
-
[45]
Xiong, R.; Sa, B.; Miao, N.; Li, Y. L.; Zhou, J.; Pan, Y.; Wen, C.; Wu, B.; Sun, Z. Structural stability and thermoelectric property optimization of Ca2Si. RSC Adv. 2017, 7, 8936−8943. doi: 10.1039/C6RA28125G
-
[46]
Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251−14269. doi: 10.1103/PhysRevB.49.14251
-
[47]
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044−2078. doi: 10.1002/jcc.21057
-
[48]
Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169−11186. doi: 10.1103/PhysRevB.54.11169
-
[49]
Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758−1775.
-
[50]
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953−17979. doi: 10.1103/PhysRevB.50.17953
-
[51]
Perdew, J. P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244−13249. doi: 10.1103/PhysRevB.45.13244
-
[52]
Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865−3868. doi: 10.1103/PhysRevLett.77.3865
-
[53]
Perdew, J. P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533−16539. doi: 10.1103/PhysRevB.54.16533
-
[54]
Monkhorst, H. J.; Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188−5192. doi: 10.1103/PhysRevB.13.5188
-
[55]
Heyd, J.; Scuseria, G. E. Efficient hybrid density functional calculations in solids: assessment of the Heyd-Scuseria-Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 2004, 121, 1187−1192. doi: 10.1063/1.1760074
-
[56]
Poater, J.; Duran, M.; Sola, M.; Silvi, B. Theoretical evaluation of electron delocalization in aromatic molecules by means of atoms in molecules (AIM) and electron localization function (ELF) topological approaches. Chem. Rev. 2005, 105, 3911−3947. doi: 10.1021/cr030085x
-
[57]
Becke, A. D.; Edgecombe, K. E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397−5403. doi: 10.1063/1.458517
-
[58]
Maintz, S.; Deringer, V. L.; Tchougréeff, A. L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557−2567. doi: 10.1002/jcc.23424
-
[59]
Wang, Y.; Chen, L. Q.; Liu, Z. K. YPHON: a package for calculating phonons of polar materials. Comput. Phys. Commun. 2014, 185, 2950−2968. doi: 10.1016/j.cpc.2014.06.023
-
[60]
Bardeen, J.; Shockley, W. S. Deformation potentials and mobilities in non-polar crystals. Phys. Rev. 1950, 80, 72−80. doi: 10.1103/PhysRev.80.72
-
[61]
Kou, L. Z.; Ma, Y. D.; Zhou, L. J.; Sun, Z. Q.; Gu, Y. T.; Du, A. J.; Smith, S.; Chen, C. F. High-mobility anisotropic transport in few-layer gamma-B-28 films. Nanoscale 2016, 8, 20111−20117. doi: 10.1039/C6NR07271B
-
[62]
Jun, D.; Cheng, Z. X. Titanium trisulfide monolayer: theoretical prediction of a new direct-gap semiconductor with high and anisotropic carrier mobility. Angew. Chem. Int. Edit. 2015, 54, 7572−7576. doi: 10.1002/anie.201502107
-
[63]
Deringer, V. L.; Zhang, W.; Rausch, P.; Mazzarello, R.; Dronskowski, R.; Wuttig, M. A chemical link between Ge-Sb-Te and In-Sb-Te phase-change materials. J. Mater. Chem. C 2015, 3, 9519−9523. doi: 10.1039/C5TC02314A
-
[64]
Deringer, V. L.; Tchougréeff, A. L.; Dronskowski, R. Crystal orbital hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 2011, 115, 5461−5466. doi: 10.1021/jp202489s
-
[65]
Qiao, J.; Kong, X.; Hu, Z. X.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475−7. doi: 10.1038/ncomms5475
-
[66]
Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147−150. doi: 10.1038/nnano.2010.279

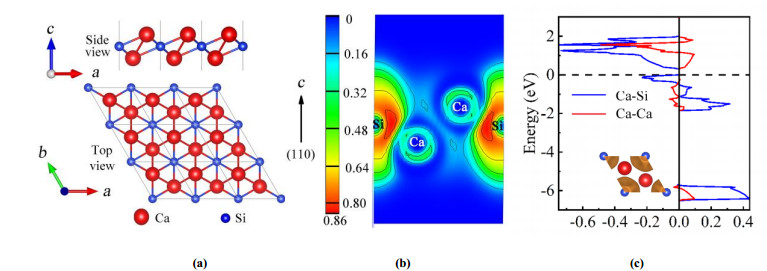
 DownLoad:
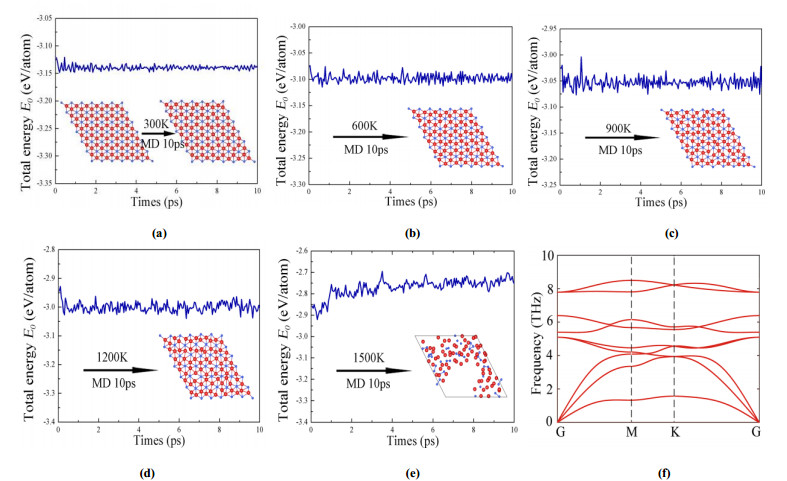
DownLoad:
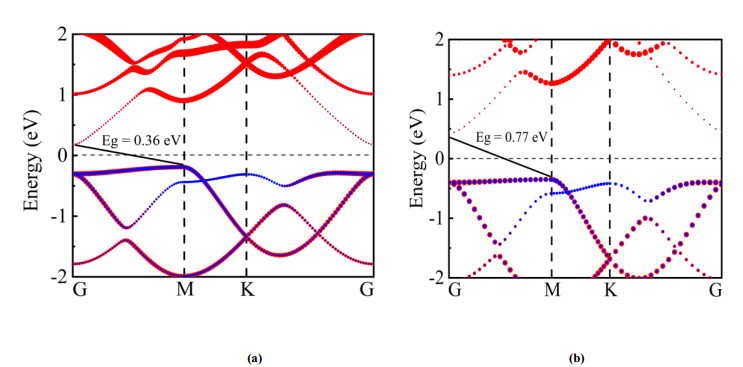
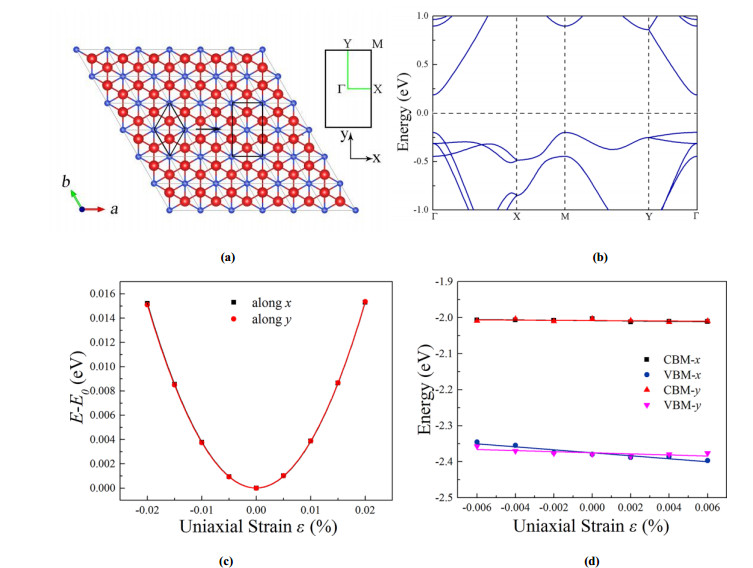

 下载:
下载:






 下载:
下载:
