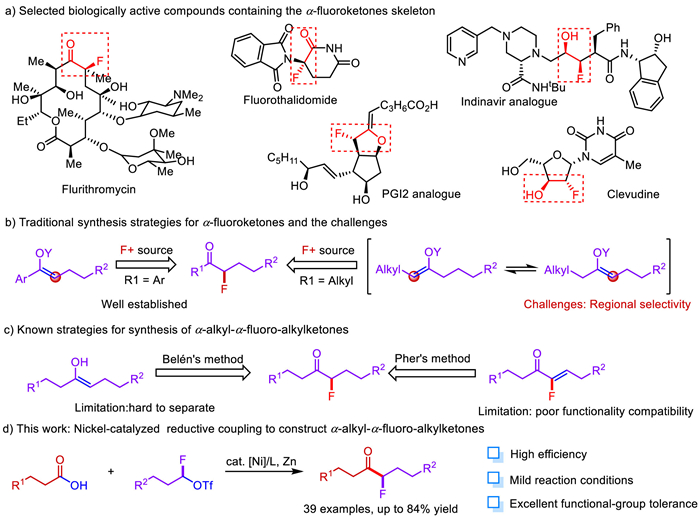
Scheme 1.
Synthesis of α-fluoroketones: Status quo & challenges.
Nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids toward the synthesis of α-alkyl-α-fluoro-alkylketones☆
Rui Wang , Jie Xu , Jin-Xiao Li , Bing-Bing Wu , Ruo-Xing Jin , Yu-Xiang Bi , Xi-Sheng Wang

As a widely applied strategy to significantly modulate the lipophilic properties, electronic, metabolic stability and bioavailability of functional molecules [1–6], the introduction of fluorine atom into organic compounds has been found widespread applications in almost all aspects of the chemical industry, ranging from pharmaceuticals, agrochemicals and materials [7–13]. However, due to the limited variety of monofluorinating agents [14–33], compared with the construction of trifluoromethyl compounds [34–46] and difluoroalkyl compounds [47–58], monofluoroalkylation especially the production of α-fluoroketones have been less intensively investigated.
α-Fluoroketones as valuable building blocks are easily found in the pharmaceutical and bioactive molecules (Scheme 1a) [59,60]. Accordingly, several methods for the synthesis of α-fluoro-arylketones have been developed via direct electrophilic/nucleophilic fluorination methods (Scheme 1b) [61–63]. The synthesis of α-fluoro-arylketones using α-fluorocarbonyl compounds as building block through the transition metals catalyzed cross-coupling was another alter alternative choice. Recently, Qing [64,65], Shreeve [66], and Wu [67] have accomplished the synthesis of α-fluoro-arylketones via palladium-catalyzed α-fluorocarbonyl compounds coupling reactions with phenylboronic acids or bromobenzenes. Moreover, Negishi and Suzuki cross-coupling of α-halo-α-fluoroketones have been shown to be effective strategies to obtain α-fluoro-arylketones [68,69]. However, to the best of our knowledge, only a few cases of directly constructing α-alkyl-α-fluoro-alkylketones have been reported. Belén has accomplished iridium-catalysed tandem isomerisation/C–F bond formation from allylic alcohols and selectfluors to prepare α-fluorinated ketones as single constitutional isomers [70]. Then Pher. G. Andersson has disclosed a straightforward method for the preparation of chiral α-alkyl-α-fluoro-alkylketones (Scheme 1c) [71]. However due to the site selectivity and substrate restriction, it still remains huge challenge to synthesize α-alkyl-α-fluoro-alkylketones easily and efficiently.
Monofluoroalkyl triflates developed by our group could be used as a leveraging modular synthetic scaffold in divergent syntheses of aliphatic monofluorides [72]. Meanwhile we have simplified the previous synthetic route in a one-step procedure by sequential addition of trifluoromethylsulfonic anhydride and Et3N·HF to the mixture of aldehydes and lutidine in dichloromethane. As expected, a series of alkyl aldehydes were smoothly transformed to the desired monofluoroalkyl triflates in a rapid and efficient fashion with high yields. To address the issues and challenges in synthesizing α-alkyl-α-fluoro-alkylketones, we envisaged monofluoroalkyl triflates as the monofluoroalkylating reagent for synthesis the desired products via nickel-catalyzed reductive coupling reactions with cheap and readily available alkyl carboxylic acids.
Herein, we describe a novel and efficient nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids, giving a series of α-alkyl-α-fluoro-alkylketones in moderate to high yields (Scheme 1d). The transformation demonstrated broad functional group compatibility and mild conditions. This method could provide a highly efficient and selective synthetic route to α-fluoroketones-containing pharmaceutical design and development.
Our initial studies commenced with 3-phenylpropionic acid (1a) as the pilot substrate, 1-fluoro-3-phenylpropyl trifluoromethanesulfonate (2a) as the mono-fluoroalkylating reagent and Boc2O (2.0 equiv.) was chosen as the activating agent to generate mixed anhydride in situ from carboxylic acids (Table 1). First, using Ni(acac)2 (10 mol%), bipyridine L1 (15 mol%), Zn (3.0 equiv.) as the reductant, MgCl2 (1.5 equiv.) as the additive and THF (0.2 mol/L) as the solvent, the desired product 3 was obtained in 14% yield (entry 1). Unfortunately, the yield of 3 could not been improved when polar solvents such as DMF, CH3CN and NMP were used (< 10%, entries 2–4). Then a series of mixed solvents were tested (for details, see Supporting information). To our delight, THF and CH3CN component solvent could afford the desired product with 38% yield (entries 5–7). To further improved the yield, various nickel catalysts were investigated, which indicated that Ni(NO3)2·6H2O was the best choice to catalyze the reaction (entries 7–11). Considered the importance of the ligand for this transformation, ligands screening was then tested. The results showed that the conversion efficiency was promoted when the electron-donating substituents were introduced on the C4-position of bipyridine, the corresponding product 3 with 62% yield could be provided by using L2 (entry 12). When L3 replacing the Me- on the ligand with more electron-donating group MeO- was investigated, the yield of 3 was enhanced to 67% (entry 13). L4 with an electron-withdrawing substituent (4-CF3) would lead to a significantly lower yield (entry 14). And higher yield could not be obtained by using phenanthroline ligands (entries 15 and 16). We then reduced TBAI to 1.2 equiv., which could further increase the yield to 75% (entry 17). It was a critical factor that TBAI and 1-fluoro-3-phenylpropyl trifluoromethanesulfonate (2a) were added after the stirring of the other reactants for 30 min. Finally, the α-alkyl-α-fluoro-alkylketone 3 could be obtained with a separation yield of 80% by step feeding (entries 18 and 19).
 DownLoad:
CSV
DownLoad:
CSV

|
With the optimized conditions established for this nickel-catalyzed reductive mono-fluoroalkylation in hand, we next started to test the substrate tolerance of this transformation (Scheme 2) First, a series of alkyl carboxylic acids were well compatible with this catalytic system for cross-coupling with 1-fluoro-3-phenylpropyl trifluoromethanesulfonate (2a). Those carboxylicacids bearing different steric properties or long chains alkyl furnished the corresponding α-alkyl-α-fluoro-alkylketone with 50%−84% yields (4–7). It should be mentioned that a series of functional groups including alkene (8, 9), chloro (10) and ester (11) were well tolerated in this catalytic method. Remarkably, the alkyl halide, alkyl ester, and olefin can serve as versatile synthetic handles for further structural elaborations. This monofluoroalkylation proceeded well with 1-naphthyl (12) and 1-pyrenyl (13) substituted alkyl carboxylic acids and gave 64%−78% yields. The substituent effect on the phenyl rings was next examined, and good yields were observed with both electron-withdrawing and electron-donating groups, such as fluoro (14), chloro (15), bromo (16), methoxy (17), 3,4-methylenedioxy (18), ester (19) and nitrile (20) were all well tolerated, which furnished the corresponding products in 43%−73% yields. To our delight, different heterocycles such as furan (21) and thiofuran (22) were also compatible and gave 78%−82% yields.
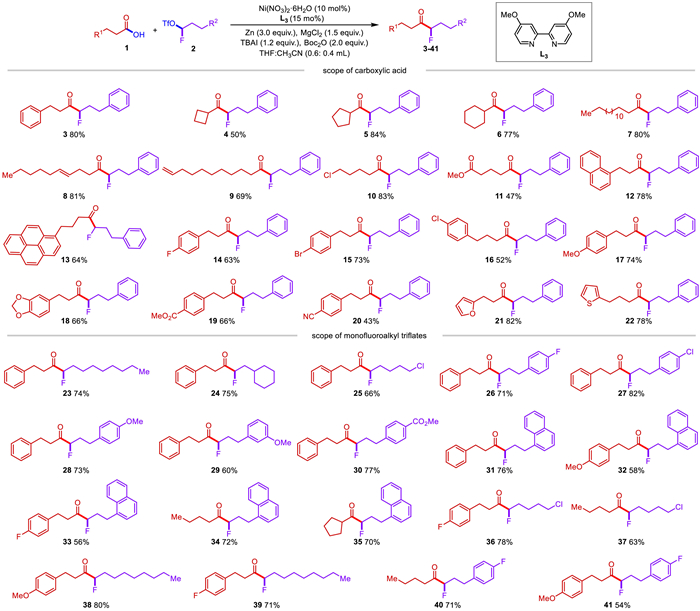
Next, we moved on to the scope of monofluoroalkylating reagents (Scheme 2). It should be noted that monofluoroalkyl triflates containing simple alkyl chains or cyclohexyl could also be successfully transformed into corresponding products with 71%−80% yields (23, 24, 38, 39). Notably, monofluoroalkyl triflates installed with a terminal chloro group on the alkyl chains were also applied onto different alkyl carboxylic acids and moderate yields of α-alkyl-α-fluoro-alkylketones were accessed (25, 36, 37). To our delight, both electron-donating groups such as methoxy (28, 29) and electron-withdrawing groups such as fluoro (26), chloro (27), ester (30) on the aryl rings were well compatible with this transformation, affording α-alkyl-α-fluoro-alkylketones with 60%−82% yields. Meanwhile, it was also suitable for the 1-naphthalene derived monofluoroalkyl triflates, which gave the desired products 31–35 in 56%−76% yields. And different alkyl carboxylic acids and monofluoroalkyl triflates can be coupled well under this strategy to obtain corresponding products (39–41).
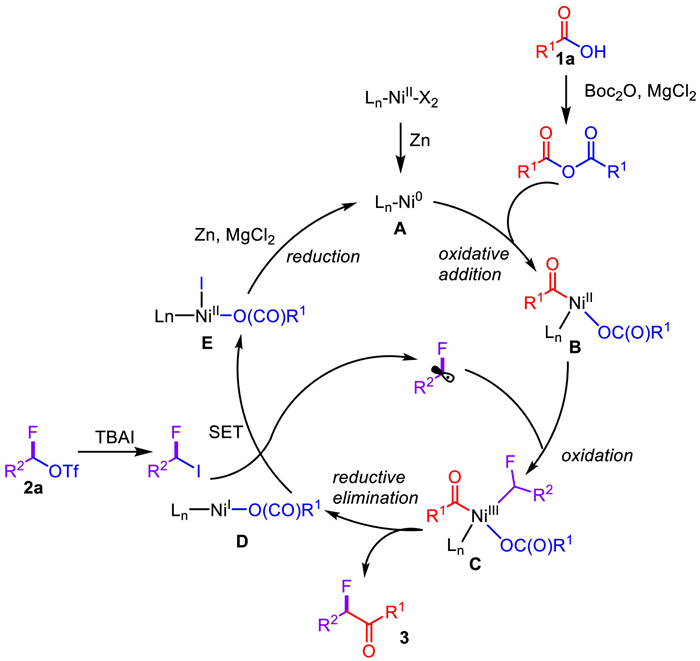
Base on the previous reports [73–75], a propose a plausible mechanism have been depicted in Scheme 3. In the presence of Zn powder, Ni(NO3)2·6H2O can be reduced to Ni(0) species A to start the catalytic process. Acid anhydride in situ formation from Boc2O and alkyl carboxylic acids followed by oxidative addition to A, giving Ni(Ⅱ) species B. Then radical oxidation to afford R1CO-LnNi(Ⅲ)-Rf (intermediate C) and subsequent afford desired α-alkyl-α-fluoro-alkylketones and R1(CO)O-LnNi(Ⅰ) species D via reductive elimination. The alkyl radical and R1(CO)O-LnNi(Ⅱ)-X can be generated by Ni(Ⅰ) species D which undergo a SET process with Rf-I. Finally, intermediate E can be reduced to Ni(0) species A by zinc powder in the presence of MgCl2 to complete the cycle.
In summary, we have developed a practical and creationary strategy for the fast synthesis of α-alkyl-α-fluoro-alkylketones. With alkyl carboxylic acids a low-cost industrial raw material, served as the acyl source, a general and efficient nickel-catalyzed reductive cross-coupling with monofluoroalkyl triflates has been established. this method demonstrated mild conditions and the excellent functional-group tolerance. α-Fluoroketones-containing pharmaceutical design and development could be completed though this efficient strategy.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge the financial support of the National Key R&D Program of China (No. 2021YFF0701700), the National Science Foundation of China (Nos. 22271264, 21971228).
Supplementary material associated with this article can be found, in the online version, at doi:
J. Wang, M. Sánchez-Roselló. J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432–2506. doi: 10.1021/cr4002879
D. O'Hagan, Chem. Soc. Rev. 37 (2008) 308–319. doi: 10.1039/B711844A
K. Mgller, C. Faeh, F. Diederich, Science 317 (2007) 1881–1886. doi: 10.1126/science.1131943
S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320–330. doi: 10.1039/B610213C
W.K. Hagmann, J. Med. Chem. 51 (2008) 4359–4369. doi: 10.1021/jm800219f
C. Ni, J. Hu, Chem. Soc. Rev. 45 (2016) 5441–5454. doi: 10.1039/C6CS00351F
P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications, 2nd Ed., Wiley-VCH, Weinheim, 2013.
M.B. van Niel, I. Collins, M.S. Beer, et al., J. Med. Chem. 42 (1999) 2087–2104. doi: 10.1021/jm981133m
R. Berger, G. Resnati, P. Metrangolo, E. Weber, J. Hulliger, Chem. Soc. Rev. 40 (2011) 3496–3508. doi: 10.1039/c0cs00221f
Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422–518. doi: 10.1021/acs.chemrev.5b00392
N.A. Meanwell, J. Med. Chem. 61 (2018) 5822–5880. doi: 10.1021/acs.jmedchem.7b01788
F. Ibba, T.D.W. Claridge, V. Gouverneur, et al., J. Am. Chem. Soc. 142 (2020) 19731–19744. doi: 10.1021/jacs.0c09832
J. He, Z. Li, G. Dhawan, et al., Chin. Chem. Lett. 34 (2023) 107578. doi: 10.1016/j.cclet.2022.06.001
Y. Zhao, B. Gao, C. Ni, J. Hu, Org. Lett. 14 (2012) 6080–6083. doi: 10.1021/ol3029737
C. Guo, X. Yue, F.L. Qing, Synthesis 11 (2010) 1837–1844.
X. Zhang, W.M. Qiu, D.J. Burton, Tetrahedron Lett. 40 (1999) 2681–2684. doi: 10.1016/S0040-4039(99)00305-6
W.K. Tang, Z.W. Xu, J. Xu, et al., Org. Lett. 21 (2019) 196–200. doi: 10.1021/acs.orglett.8b03656
Y. Zhao, C. Ni, F. Jiang, et al., ACS Catal. 3 (2013) 631–634. doi: 10.1021/cs4000574
X. Jiang, S. Sakthivel, K. Kulbitski, G. Nisnevich, M. Gandelman, J. Am. Chem. Soc. 136 (2014) 9548–9551. doi: 10.1021/ja504089y
J. Hu, B. Gao, L. Li, C. Ni, J. Hu, Org. Lett. 17 (2015) 3086–3089. doi: 10.1021/acs.orglett.5b01361
Y.M. Su, G.S. Feng, Z.Y. Wang, Q. Lan, X.S. Wang, Angew. Chem. Int. Ed. 54 (2015) 6003–6007. doi: 10.1002/anie.201412026
L. An, Y.L. Xiao, Q.Q. Min, X. Zhang, Angew. Chem. Int. Ed. 54 (2015) 9079–9083. doi: 10.1002/anie.201502882
Y. Wu, H.R. Zhang, Y.X. Cao, Q. Lan, X.S. Wang, Org. Lett. 18 (2016) 5564–5567. doi: 10.1021/acs.orglett.6b02803
J. Sheng, H.Q. Ni, G. Liu, Y. Li, X.S. Wang, Org. Lett. 19 (2017) 4480–4483. doi: 10.1021/acs.orglett.7b02012
N.Y. Wu, X.H. Xu, F.L. Qing, ACS Catal. 9 (2019) 5726–5731. doi: 10.1021/acscatal.9b01530
H. Yin, J. Sheng, K.F. Zhang, et al., Chem. Commun. 55 (2019) 7635–7638. doi: 10.1039/c9cc03737c
N.A. Beare, J.F. Hartwig, J. Org. Chem. 67 (2002) 541–555. doi: 10.1021/jo016226h
E. Cosimi, J. Saadi, H. Wennemers, Org. Lett. 18 (2016) 6014–6017. doi: 10.1021/acs.orglett.6b02795
J. Sheng, H.Q. Ni, S.X. Ni, et al., Angew. Chem. Int. Ed. 60 (2021) 15020–15027. doi: 10.1002/anie.202102481
S.X. Ni, Y.L. Li, H.Q. Ni, et al., Chin. Chem. Lett. 34 (2023) 107614. doi: 10.1016/j.cclet.2022.06.037
F.L. Qing, X.Y. Liu, J.A. Ma, et al., CCS Chem. 4 (2022) 2518–2549. doi: 10.31635/ccschem.022.202201935
Y. Li, W. Liu, Z.Y. Liu, et al., CCS Chem. 4 (2022) 2888–2896. doi: 10.31635/ccschem.021.202101395
Y. Hu, J. Luo, C. Lü, Chin. Chem. Lett. 21 (2010) 151–154. doi: 10.1016/j.cclet.2009.10.014
Y.Z. Cheng, J. Ma, Y. Zhang, S.Y. Yu, Adv. Synth. Catal. 356 (2014) 2859–2866. doi: 10.1002/adsc.201400504
L. Li, Q.Y. Chen, Y. Guo, J. Fluorine Chem. 167 (2014) 79–83.
Y.Y. Yu, G.I. Georg, Adv. Synth. Catal. 356 (2014) 3510–3518. doi: 10.1002/adsc.201400417
B. Sahoo, J.L. Li, F. Glorius, Angew. Chem. Int. Ed. 54 (2015) 11577–11580. doi: 10.1002/anie.201503210
R. Tomita, T. Koike, M. Akita, Angew. Chem. Int. Ed. 54 (2015) 12923–12927. doi: 10.1002/anie.201505550
N. Noto, T. Koike, M. Akita, J. Org. Chem. 81 (2016) 7064–7071. doi: 10.1021/acs.joc.6b00953
J.S. Lin, B. Tan, X.Y. Liu, J. Am. Chem. Soc. 138 (2016) 9357–9360. doi: 10.1021/jacs.6b04077
H. Xiao, Z. Liu, H. Shen, et al., Chem 5 (2019) 940–949. doi: 10.1016/j.chempr.2019.02.006
H. Wang, Y. Xie, Y. Zhou, N. Cen, W. Chen, Chin. Chem. Lett. 33 (2022) 221–224. doi: 10.1016/j.cclet.2021.06.008
B.B. Wu, J. Xu, K.J. Bian, Q. Gao, X.S. Wang, J. Am. Chem. Soc. 144 (2022) 6543–6550. doi: 10.1021/jacs.2c01422
H. Luo, Y. Zhao, D. Wang, M. Wang, Z. Shi, Green Synth. Catal. 1 (2020) 134–142. doi: 10.1016/j.gresc.2020.08.002
Y. Dai, F. Wang, S. Zhu, L. Chu, Chin. Chem. Lett. 33 (2022) 4074–4078. doi: 10.1016/j.cclet.2021.12.050
N. Meng, Y. Lv, X. Zhao, W. Wei, Chin. Chem. Lett. 32 (2021) 258–262. doi: 10.1016/j.cclet.2020.11.034
Z. Feng, Q.Q. Min, Y.L. Xiao, B. Zhang, X. Zhang, Angew. Chem. Int. Ed. 53 (2014) 1669–1673. doi: 10.1002/anie.201309535
M.K. Schwaebe, J.R. McCarthy, J.P. Whitten, Tetrahedron Lett. 41 (2000) 791–794. doi: 10.1016/S0040-4039(99)02212-1
Y.J. Chen, L.K. Li, Y.Y. Ma, Z.P. Li, J. Org. Chem. 84 (2019) 5328–5338. doi: 10.1021/acs.joc.9b00339
X. Huang, Y. Zhang, C. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 5956–5961. doi: 10.1002/anie.201900745
H.Y. Zhao, X. Gao, S. Zhang, X. Zhang, Org. Lett. 21 (2019) 1031–1036. doi: 10.1021/acs.orglett.8b04070
H. Chen, J. Wang, J. Wu, Y. Kuang, F. Wu, J. Fluorine Chem. 200 (2017) 41–46. doi: 10.1016/j.jfluchem.2017.06.003
J.X. Wang, J.J. Wu, H. Chen, S.W. Zhang, F.H. Wu, Chin. Chem. Lett. 26 (2015) 1381–1384. doi: 10.1016/j.cclet.2015.07.007
D. Wang, J. Wu, J. Huang, et al., Tetrahedron 73 (2017) 3478–3484. doi: 10.1016/j.tet.2017.05.021
J. Liang, G. Huang, P. Peng, et al., Adv. Synth. Catal. 360 (2018) 2221–2227. doi: 10.1002/adsc.201701569
X. Chen, Z. Zhu, S. Liu, Y.H. Chen, X. Shen, Chin. Chem. Lett. 33 (2022) 2391–2396. doi: 10.1016/j.cclet.2021.10.083
W. Wu, Y. You, Z. Weng, Chin. Chem. Lett. 33 (2022) 4517–4530. doi: 10.1016/j.cclet.2022.01.038
J. Li, W. Xi, S. Liu, et al., Chin. Chem. Lett. 33 (2022) 3007–3011. doi: 10.1016/j.cclet.2021.12.002
S.D. Putnam, M. Castanheira, G.J. Moet, D.J. Farrell, R.N. Jones, Diagn. Microbiol. Infect. Dis. 66 (2010) 393–401. doi: 10.1016/j.diagmicrobio.2009.10.013
P.A. Champagne, J.F. Paquin, Chem. Rev. 115 (2015) 9073–9174. doi: 10.1021/cr500706a
T. Ishimaru, N. Shibata, T. Horikawa, et al., Angew. Chem. Int. Ed. 47 (2008) 4157–4161. doi: 10.1002/anie.200800717
H. Teare, E.G. Robins, E. Arstad, S.K. Luthra, V. Gouverneur, Chem. Commun. ˚ (2007) 2330–2332.
W. Zhang, J. Hu, Adv. Synth. Catal. 352 (2010) 2799–2804. doi: 10.1002/adsc.201000499
C. Guo, R.W. Wang, Y. Guo, F.L. Qing, J. Fluorine Chem. 133 (2012) 86–96. doi: 10.1016/j.jfluchem.2011.08.004
C. Guo, R.W. Wang, F.L. Qing, J. Fluorine Chem. 143 (2012) 135–142. doi: 10.1016/j.jfluchem.2012.05.001
Y. Guo, B. Twamley, J.M. Shreeve, Org. Biomol. Chem. 7 (2009) 1716–1722. doi: 10.1039/b900311h
J. Zhou, X. Fang, T.L. Shao, X.Y. Yang, F.H. Wu, J. Fluorine Chem. 191 (2016) 54–62. doi: 10.1016/j.jfluchem.2016.09.016
Y.F. Liang, G.C. Fu, J. Am. Chem. Soc. 136 (2014) 5520–5524. doi: 10.1021/ja501815p
J. Liang, J. Han, J. Wu, et al., Org. Lett. 21 (2019) 6844–6849. doi: 10.1021/acs.orglett.9b02474
N. Ahlsten, B. Martín-Matute, Chem. Commun. 47 (2011) 8331–8333. doi: 10.1039/c1cc12653a
S. Ponra, J. Yang, S. Kerdphon, P.G. Andersson, Angew. Chem. Int. Ed. 58 (2019) 9282–9287. doi: 10.1002/anie.201903954
B.B. Wu, J. Xu, Q. Gao, et al., Angew. Chem. Int. Ed. 61 (2022) e202208938. doi: 10.1002/anie.202208938
C. Zhao, X. Jia, X. Wang, H. Gong, J. Am. Chem. Soc. 136 (2014) 17645–17651. doi: 10.1021/ja510653n
J.B. Diccianni, T. Diao, Trends Chem. 1 (2019) 830–844. doi: 10.1016/j.trechm.2019.08.004
C.S. Day, Á. Rentería-Gómez, S.J. Ton, et al., Nat. Catal. 6 (2023) 244–253. doi: 10.1038/s41929-023-00925-4

Scheme 2 Scope of nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids. Reaction conditions were as follows: 1 (0.2 mmol), 2 (0.3 mmol), [Ni] (10 mol%), L3 (15 mol%), TBAI (1.2 equiv.), Boc2O (0.4 mmol, 2.0 equiv.), MgCl2 (0.3 mmol, 1.5 equiv.), Zn (0.6 mmol, 3.0 equiv.), THF: CH3CN (0.6 mL: 0.4 mL), 35 ℃, 12 h.
Table 1. Optimization of reaction conditions of nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids. a
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: