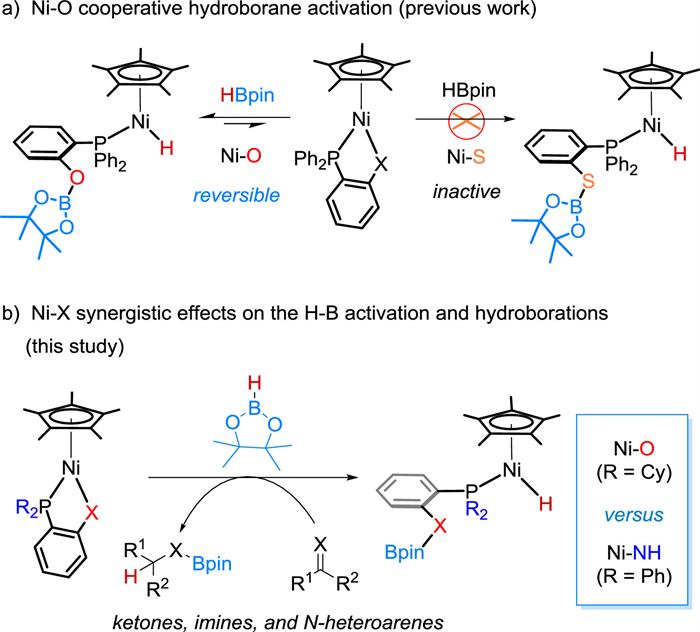
Scheme 1.
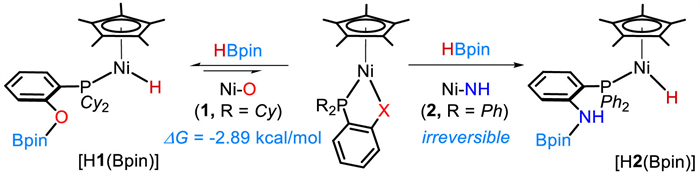
Cooperative activation of HBpin by Cp*Ni(1,2-R2PC6H4X) complexes.

Cooperative metal-ligand reactivity for bond activation and catalysis is a powerful strategy in the development of homogenous catalysts [1-7]. Significantly, it can widely expand the use of base metal catalysts for sustainable transformations and organic synthesis [8-17]. During the renaissance of nickel catalysis over recent years [18,19], the ligand scaffolds were well designed and operated in a synergistic fashion with the metal to participate in reaction sequences and serving as a Lewis base [20-30], an electron reservoir [31-33] or through dearomatization-aromatization [34-36]. These studies have promoted nickel catalysis in many important applications [34-50], such as hydroboration of CO2 [13], production and oxidation of H2 [51-54], and hydrosilylation of alkenes [31,32,34].
Our group has explored metal-ligand cooperation (MLC) chemistry on a Cp*M(1,2-Ph2PC6H4X) (M = Fe, Co or Ni; X = S, HN or O) platform. By switching the M-X pair and modifying the electronic and steric factors at the phosphorus, a new class of half-sandwich complexes has been developed and used in a multitude of catalytic reactions [55-62]. These M-X complexes have shown intriguing reactivity in activation of hydroboranes [57], ammonia-borane [58], epoxides [59] and terminal alkynes [60,61]. In studies of cooperative B-H bond activation of HBpin, we found that the reactivity of Cp*M(1,2-Ph2PC6H4X) is sensitive to the M-X pair, which also affects catalytic performance of the complex. For example, the Fe−S complex, Cp*Fe(1,2-Ph2PC6H4S), is inert toward HBpin but can activate aryl epoxides for their hydroboration [59]. In contrast, the Ni−O complex, Cp*Ni(1,2-Ph2PC6H4O), achieves activation of HBpin at its Ni−O bond [62].
When the anionic coordination site is changed however from oxygen to sulfur, Cp*Ni(1,2-Ph2PC6H4S), regains its stability toward HBpin (Scheme 1a).
Nickel-based cooperative B-H bond activation is attractive because it allows direct use of HBpin rather than exogenous activators to generate Ni(Ⅱ)−H species for catalytic hydro-boration reactions. In the Cp*Ni(1,2-R2PC6H4X) half-sandwich system, the parent nickel complex and the resulting hydride intermediate both have an 18-electron configuration, and are stable in air. The challenge to achieve such MLC catalysis is not only activation of the B-H bond but also generation of a sufficiently active hydride intermediate, HNi—X(Bpin) that allows the hydro-boration sequences to proceed [15,62].
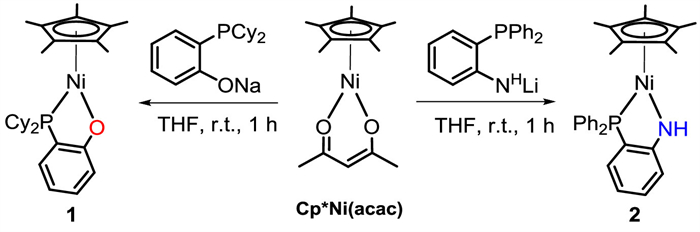
Building on our previous work with the Ni−O complex, we aimed to investigate the synergistic effects of Ni−X on cooperative B-H bond activation and the catalytic performance in hydroboration reactions associated with subtle tuning of the Ni−X and the ancillary phosphine. Herein, we describe two new nickel complexes Cp*Ni(1,2-Cy2PC6H4O) (1) and Cp*Ni(1,2-Ph2PC6H4NH) (2) which participate in cooperative B-H bond activation, and report their catalytic performance in hydroboration of unsaturated organic compounds containing C=X groups (Scheme 1b).
According to a previously reported method [62], complexes 1 and 2 were synthesized independently by treating a solution in THF of Cp*Ni(acac) with equimolar amounts of 1,2-Cy2PC6H4ONa or 1,2-Ph2PC6H4NHLi (Scheme 2). Complexes 1 and 2 were isolated as air-stable yellow solids in yields of 90%–94%. In their 31P NMR spectra, the C6D6 solution of the complexes have a sharp singlet at δ 41.82 for 1 and 45.07 for 2. The solid-state structures of 1 and 2 were confirmed by X-ray crystallography (Fig. 1). They both exhibit a framework similar to that of Cp*Ni(1,2-R2PC6H4X), which adopts a two-legged piano-stool geometry. The Ni-P bond lengths in 1 (2.1558(7) Å) and 2 (2.1382(6) Å) are very close to those reported for Cp*Ni(1,2-Ph2PC6H4O) (2.1290(1) Å) and Cp*Ni(1,2-Ph2PC6H4S) (2.1140(1) Å) [62].
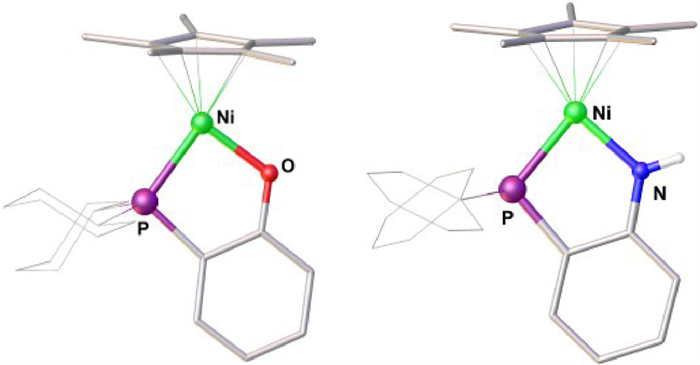
The redox properties of 1 and 2 were investigated by cyclic voltammetry (Fig. 2a). They both exhibit two reversible redox events, which were tentatively assigned to the Ni(Ⅱ)/Ni(Ⅲ) and Ni(Ⅰ)/Ni(Ⅱ)) couples. The oxidation potential of 1+/0 was observed as 0.14 V vs. Fc+/0, about 0.26 V less negative than that of 2+/0 (−0.12 V vs. Fc+/0). However, their reduction potentials −1.96 V for 10/- versus −2.02 V for 20/- differ only slightly. These results indicate that the metal center of the Ni−N complex is more electron-rich compared with that in the Ni−O complex.
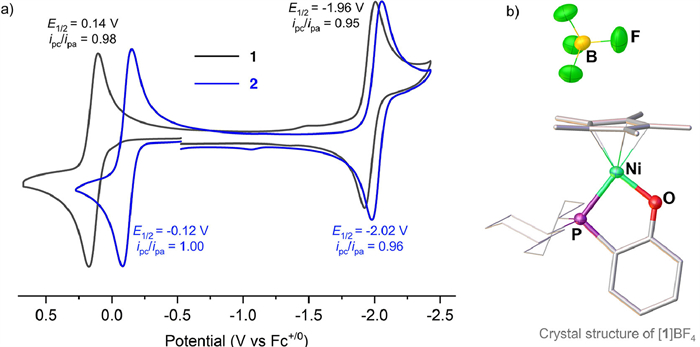
At −30 ℃, oxidation of 1 by AgBF4 in CH2Cl2 was conducted on a preparative scale. The resulting cationic Ni(Ⅲ) complex, was isolated in 97% yield, and its structure was successfully determined by X-ray crystallography (Fig. 2b). Although the framework of 1+ in [1]BF4 is almost identical to that of 1, some bond lengths and angles are slightly different. For example, the oxidation leads to an increase in the Ni−P distance from 2.1558(7) Å in 1 to 2.2307(6) Å in [1]+, while the Ni−O distance decreased from 1.886(2) Å to 1.8229(15) Å.
Both 1 and 2 react with HBpin to afford a Ni(Ⅱ)−H species, but show different reactivities in activation of the B-H bond (Scheme 3). The reaction of 1 with 4 equiv. of HBpin in C6D6 was conducted in the J. Young NMR tube and monitored by NMR spectroscopy. After 3 h, the 31P NMR spectroscopic studies indicated that 1 had been completely converted to a new hydride species [H1(Bpin)] which has a 31P signal at δ 71.72. In the 1H NMR spectra, a characteristic hydride resonance was displayed at δ −22.34 as a doublet (d, J = 103.2 Hz). When 1.2 equiv. of HBpin was used, we found that 1 cannot be completely consumed even after reacting for 48 h. The reaction ultimately provided an equilibrium mixture of 1 and [H1(Bpin)] in a ratio of approximately 3.62:1.00 as determined by the 31P NMR spectrum, revealing a calculated equilibrium constant (Keq) of 131.9. These experimental observations indicate that the activation at the Ni-O complex is reversible [62]. The free energy change for the reaction of 1 with HBpin was calculated based on the equilibrium constant, as −2.89 kcal/mol at 298 K.
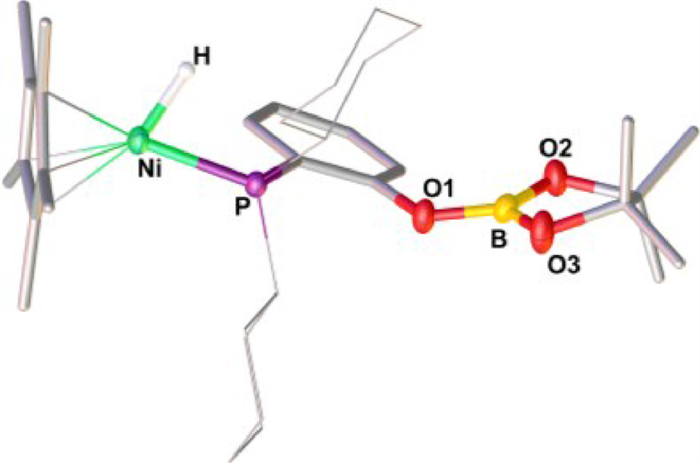
Slow diffusion of hexane into the solution of 1 reacting with excess HBpin at −30 ℃ provided brown-red crystals of [H1(Bpin)], some of which were suitable for X-ray single crystal diffraction. Crystallographic analysis of [H1(Bpin)] confirmed the solid-sate structure of a neutral nickel hydride, adopting a piano-stool type, 18-electron configuration (Fig. 3). Judging from the structure, the B-H bond of HBpin was added across the Ni−O bond to provide a Ni(Ⅱ)−H bond. The O atom is connected to the boryl moiety but detached from the metal center. The hydride position was located and the length of the Ni−H bond was found to be 1.42(2) Å, in the range of 1.32–1.65 Å reported for the Cp*NiH(PR3) systems [18,62,63].
Unlike the Ni−O complex (1), the Ni−NH complex (2) reacts irreversibly with HBpin. With the stoichiometric reaction of 2 with HBpin at room temperature (r.t.) for 1 h, NMR studies indicated that 2 was cleanly converted to the nickel(Ⅱ) hydride, [H2(Bpin)]. Only one 31P NMR signal was observed at δ 38.99 and in the 1H NMR spectrum, a hydride signal showed at δ −19.81 as a doublet with JP−H = 97.9 Hz. The 11B NMR spectrum displayed a broad signal at δ 23.4, while 11B coupling with the hydride nuclei was not observed, indicative of a complete cleavage of the B-H bond. Upon redissolving the solid of [H2(Bpin)] in C6D6, the 31P NMR spectrum continued to display only one signal at 38.99 for [H2(Bpin)], and the formation of 2 was not observed within 24 h at r.t. These studies indicate the addition of HBpin to this Ni−N system is irreversible.
In view of the activation of HBpin, we subsequently evaluated the catalytic performance of 1 and 2 in hydroboration of ketones and imines (Scheme 4). The hydroboration reactions were performed with 1.5 equiv. of HBpin and 2 mol% catalyst loading in C6D6 at r.t. Complex 1 showed high catalytic activity in the hydroboration of ketones, while 2 was found to be less active. In the presence of 2 mol% of 1, acetophenone, 4-methoxyacetophenone and benzophenone all underwent smooth hydroboration, and were reduced to the corresponding boronate esters (4a-4c) nearly quantitatively within 3 h. When 2 was used as a catalyst, the hydroboration reactions proceeded much more slowly. The reaction of 4-methoxyacetophenone with HBpin for 3 h for example under the catalytic conditions produced 4b in only 18% yield. With 2 mol% of 2, the reaction of benzophenone with HBpin for 8 h produced the boronic ester (4c) in 53% yield, compared to the 99% yield of 4c that was obtained within 3 h using the catalyst (1). Interestingly, 2-phenylcyclohexan-1-one (4d), a cyclic alkyl-substituted ketone, can be efficiently hydroborated in a reasonable reaction time, using either 1 or 2 as the catalyst.
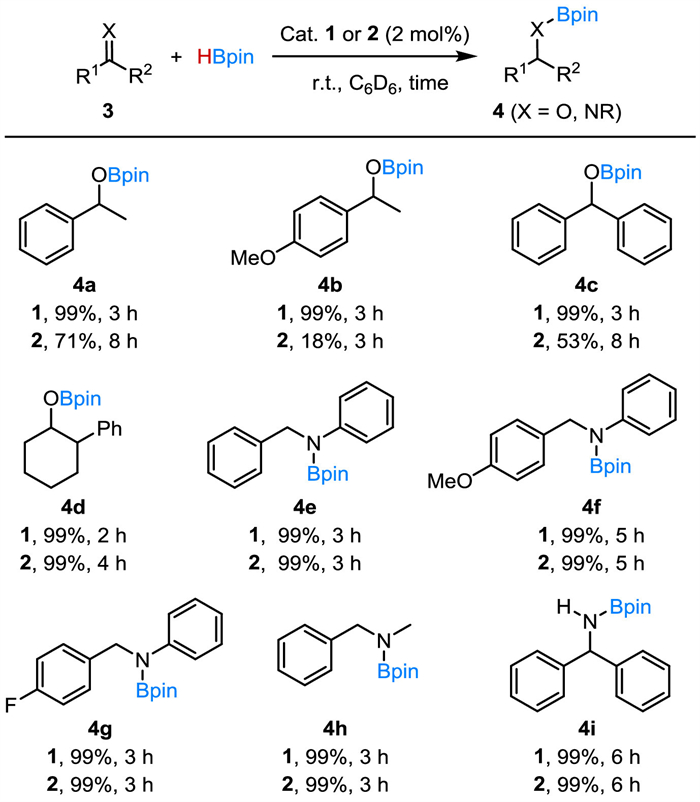
Complexes 1 and 2 both are efficient catalysts in the hydroboration of imines. Under the catalytic conditions, N-benzylideneaniline and its derivatives with electron-donating or electron-withdrawing substituents at the para-position of the benzyl group all allowed for the hydroboration producing the desired borylated amines (4e-4g) in excellent yields. The hydroboration was not affected by varying the substituents on the imine nitrogen atom. At a 2 mol% catalyst loading, ketimines such as N-alkylimine and diphenylmethanimine were completely hydro-borated to the amine products (4h, 4i) within 3 h.
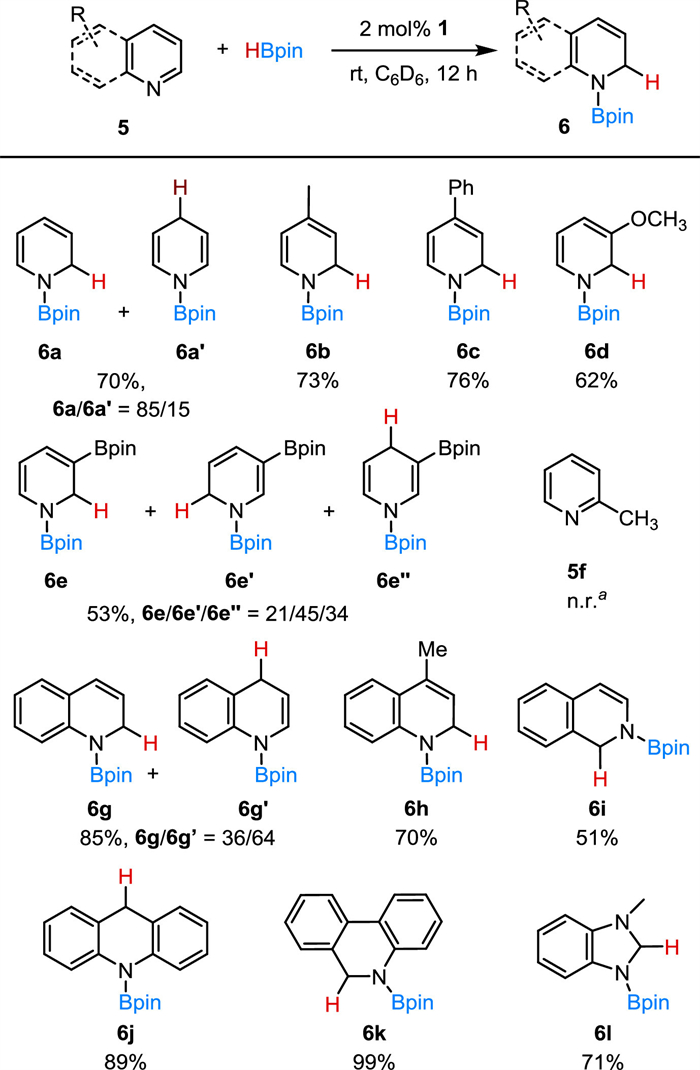
Catalytic selective reduction of N-heterocycles through hydrosilylation or hydroboration is particularly interesting, because it provides valuable dihydropyridine compounds [58,64-69]. The related transformation using nickel-based catalysis has been reported only rarely [13,14,62]. In this way, we subsequently evaluated the catalytic performances of the half-sandwich nickel complexes on the hydroboration of pyridines. Compared to 1, a greatly diminished catalytic activity was shown by 2, and was demonstrated by hydroboration of 4-methyl pyridine (5b) (Eq. 1).
|
|
(1) |
On the basis of the pyridine hydroboration catalyzed by 1, we investigated the substrate scope for this nickel-catalyzed dearomatization reaction (Scheme 5). Upon 1,2-hydroboration para-substituted pyridines afforded the dihydropyridine products (6b, 6c). Hydroboration of 3-methoxypyridine (6d) was proved to be difficult for the Lewis acid B(Me)Ar2F catalysis [70]. With the present Ni-catalyzed protocol, 5d was regioselectively hydroborated to the 1,2-product in a moderate yield (6d, 62%). The electronic and steric factors at the N-heterocycle ring appear to affect the regioselectivity of the outcome. For the substrate with a meta-substituted -Bpin functional group (5e), the reaction produced a mixture of the 1,2-, 1,4- and 1,6-dearomatized isomers in a ratio of 6e/6e′/6e″ = 21/45/34. Probably as a result of the steric hindrance, ortho-substituted pyridines such as 2-methylpyridine (5f) are not suitable for the hydroboration. This Ni−O system exhibits good activity in reduction of benzo fused N-heterocycles. Hydroboration of quinoline gave a mixture of 1,2-and 1,4-products (6h/6h′ = 36/64) in 85% yield. When a methyl group was introduced at the para-position of quinoline, as in 4-methylquinoline (5i), the 1,2-regiospecific reduction was achieved exclusively. Other N-heteroarenes such as isoquinoline (5j), acridine (5k), phenanthridine (5l) and methyl-1H-benzo[d]-imidazole (5m) all underwent regioselective hydroboration smoothly to provide the dearomatized dihydropyridine analogues (6j-6m) with good to excellent yields.
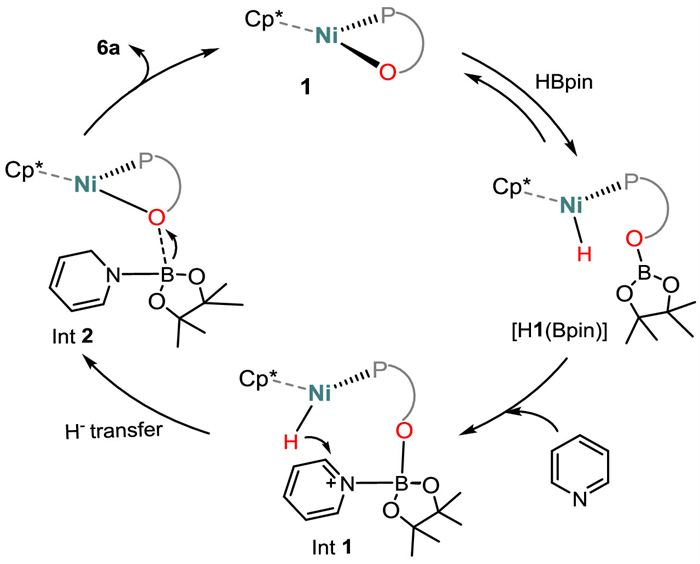
Combining our previous studies of the mechanism [62,69] with the stoichiometric HBpin activation by 1, we propose that the catalytic pyridine hydroboration is initiated by formation of [H1(Bpin)] through addition of the B-H bond across the Ni−O bond (Scheme 6). The resulting H—Ni(Ⅱ)—O(Bpin) species interacts with pyridine by coordination of the substrate with the boron atom of the Bpin moiety (Int1), and this facilitates the transfer of hydride from Ni(Ⅱ)-H to the ortho-carbon of the pyridine ring (Int2). Once the pyridine ring is dearomatized, the hydroboration can be accomplished by the cleavage of the O-B bond to release the 1,2-product, recovering the Ni−O catalyst.
In conclusion, we have demonstrated cooperative B-H bond activation in HBpin by half-sandwich Ni−O and Ni−N complexes which feature anionic coordination sites, and their participation in the catalytic hydroboration of unsaturated organic compounds. The reactions involve addition of the B-H bond across the Ni−X bond, giving rise to H—Ni(Ⅱ)—X(Bpin) hydride intermediates with an anionic site X-stabilized boron moiety. Notably, subtle tuning of the Ni−X pair and support for the ancillary phosphine significantly affects the reactivity and catalytic performance of Cp*Ni(1,2-R2PC6H4X).
The reversible activation of HBpin by the Ni-O complex is essential to the catalytic hydroboration of N-heteroarenes, probably because activation of the N-heterocyclic ring in some degree by coordination with the Bpin moiety of H—Ni(Ⅱ)—O(Bpin) is necessary for completion of the subsequent hydride reduction step. In the case of H—Ni(Ⅱ)—N(Bpin), the irreversible binding of a Bpin moiety at the nitrogen site makes its interaction with and activation of a pyridine substrate unfavorable, resulting in low catalytic activity for the hydroboration. It is less likely that the hydroboration of ketones and imines by the Ni−X complexes might proceed through a different mechanism, i.e., transfer of the hydride from H—Ni(Ⅱ)—X(Bpin) to the C=X group is prior to the Bpin transfer [71,72].
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
W. Wang would like to thank the financial support from the National Natural Science Foundation of China (Nos. 22022102 and 22071010). J. Liu gratefully acknowledges the financial support from China Postdoctoral Science Foundation (No. 2021M700462).
H. Grützmacher, Angew. Chem. Int. Ed. 47 (2008) 1814–1818. doi: 10.1002/anie.200704654
C. Gunanathan, D. Milstein, Acc. Chem. Res. 44 (2011) 588–602. doi: 10.1021/ar2000265
M. Zafar, A. Ahmad, S. Saha, et al., Chem. Sci. 13 (2022) 8567–8575. doi: 10.1039/d2sc00907b
M. Stradiotto, R.J. Lundgren, Ligand Design in Metal Chemistry: Reactivity and Catalysis, Wiley Online Library, 2016.
L. Omann, C.D.F. Königs, H.F.T. Klare, et al., Acc. Chem. Res. 50 (2017) 1258–1269. doi: 10.1021/acs.accounts.7b00089
K.D. Spielvogel, N.C. Stumme, T.V. Fetrow, et al., Inorg. Chem. 61 (2022) 2391–2401. doi: 10.1021/acs.inorgchem.1c03014
X. Sun, X. Gong, Z. Xie, et al., Chin. J. Chem. 40 (2022) 2047–2053. doi: 10.1002/cjoc.202200176
J.I. van der Vlugt, Eur. J. Inorg. Chem. 3 (2012) 363–375. doi: 10.1002/ejic.201100752
W. Yang, Z. Yang, L. Chen, et al., Chin. Chem. Lett. 34 (2023) 107791. doi: 10.1016/j.cclet.2022.107791
Y. Wang, Y. He, S. Zhu, Acc. Chem. Res. 55 (2022) 3519–3536. doi: 10.1021/acs.accounts.2c00628
R.H. Morris, Acc. Chem. Res. 48 (2015) 1494–1502. doi: 10.1021/acs.accounts.5b00045
T. Zell, D. Milstein, Acc. Chem. Res. 48 (2015) 1979–1994. doi: 10.1021/acs.accounts.5b00027
S. Chakraborty, P. Bhattacharya, H. Dai, et al., Acc. Chem. Res. 48 (2015) 1995–2003. doi: 10.1021/acs.accounts.5b00055
X.Y. Liu, H.-B. Zhu, Y.J. Shen, et al., Chin. Chem. Lett. 28 (2017) 350–353. doi: 10.1016/j.cclet.2016.09.006
Q. Liang, D. Song, Chem. Soc. Rev. 49 (2020) 1209–1232. doi: 10.1039/c9cs00508k
Y. Zhao, X. Yu, H. Hu, et al., Chin. Chem. Lett. 29 (2018) 1651–655. doi: 10.1016/j.cclet.2018.03.013
J. Liu, A. Zhang, H. Song, et al., Chin. Chem. Lett. 29 (2018) 949–953. doi: 10.1016/j.cclet.2017.09.059
N.A. Eberhardt, H. Guan, Chem. Rev. 116 (2016) 8373–8426. doi: 10.1021/acs.chemrev.6b00259
S.R. Tamang, A. Singh, D.K. Unruh, et al., ACS Catal. 8 (2018) 6186–6191. doi: 10.1021/acscatal.8b01166
Z.R. Dong, Y.Y. Li, S.L. Yu, et al., Chin. Chem. Lett. 23 (2012) 533–536. doi: 10.1016/j.cclet.2012.02.005
Z.K. Sweeney, J.L. Polse, R.G. Bergman, et al., Organometallics 18 (1999) 5502–5510. doi: 10.1021/om9907876
D. Sellmann, F. Geipel, M. Moll, Angew. Chem. Int. Ed. 39 (2000) 561–563. doi: 10.1002/(SICI)1521-3773(20000204)39:3<561::AID-ANIE561>3.0.CO;2-3
F.-G. Fontaine, D. Zargarian, J. Am. Chem. Soc. 126 (2004) 8786–8794. doi: 10.1021/ja048911m
X. Yang, M.B. Hall, J. Am. Chem. Soc. 130 (2008) 1798–1799. doi: 10.1021/ja0751328
W.H. Harman, J.C. Peters, J. Am. Chem. Soc. 134 (2012) 5080–5082. doi: 10.1021/ja211419t
W.H. Harman, T.P. Lin, J.C. Peters, Angew. Chem. Int. Ed. 53 (2014) 1081–1086. doi: 10.1002/anie.201308175
D.V. Gutsulyak, W.E. Piers, J. Borau-Garcia, et al., J. Am. Chem. Soc. 135 (2013) 11776–11779. doi: 10.1021/ja406742n
E.A. LaPierre, W.E. Piers, D.M. Spasyuk, et al., Chem. Commun. 52 (2016) 1361–1364. doi: 10.1039/C5CC09349J
F. Olechnowicz, G.L. Hillhouse, T.R. Cundari, et al., Inorg. Chem. 56 (2017) 9922–9930. doi: 10.1021/acs.inorgchem.7b01420
L.T. Scharf, A. Kowsari, T. Scherpf, et al., Organometallics 38 (2019) 4093–4104. doi: 10.1021/acs.organomet.9b00386
T.J. Steiman, C. Uyeda, J. Am. Chem. Soc. 137 (2015) 6104–6110. doi: 10.1021/jacs.5b03092
I. Pappas, S. Treacy, P.J. Chirik, ACS Catal. 6 (2016) 4105–4109. doi: 10.1021/acscatal.6b01134
C.L. Rock, T.L. Groy, R.J. Trovitch, Dalton Trans. 47 (2018) 8807–8816. doi: 10.1039/C8DT01857J
G. Vijaykumar, A. Pariyar, J. Ahmed, et al., Chem. Sci. 9 (2018) 2817–2825. doi: 10.1039/C7SC04687A
D. Oren, Y. Diskin-Posner, L. Avram, et al., Organometallics 37 (2018) 2217–2221. doi: 10.1021/acs.organomet.8b00160
M.R. Elsby, R.T. Baker, Chem. Soc. Rev. 49 (2020) 8933–8987. doi: 10.1039/d0cs00509f
S. Chakraborty, J.A. Krause, H. Guan, Organometallics 28 (2009) 582–586. doi: 10.1021/om800948f
J. Zhang, C.M. Medley, J.A. Krause, et al., Organometallics 29 (2010) 6393–6401. doi: 10.1021/om100816d
S. Chakraborty, Y.J. Patel, J.A. Krause, et al., Angew. Chem. Int. Ed. 52 (2013) 7523–7526. doi: 10.1002/anie.201302613
T.J. Schmeier, N. Hazari, C.D. Incarvito, et al., Chem. Commun. 47 (2011) 1824–1826. doi: 10.1039/C0CC03898A
L. Fan, O.V. Ozerov, Chem. Commun. (2005) 4450–4452. doi: 10.1039/b505778g
Z. Wu, T. Si, G. Xu, et al., Chin. Chem. Lett. 30 (2019) 597–600. doi: 10.1016/j.cclet.2018.12.027
C. Sun, G. Yin, Chin. Chem. Lett. 33 (2022) 5096–5100. doi: 10.1016/j.cclet.2022.04.026
Y. He, Y. Cai, S. Zhu, J. Am. Chem. Soc. 139 (2017) 1061–1064. doi: 10.1021/jacs.6b11962
Y. He, J. Ma, H. Song, et al., Nat. Commun. 13 (2022) 2471. doi: 10.1038/s41467-022-30006-2
J. Xiao, Y. He, F. Ye, et al., Chem 4 (2018) 1645–1657. doi: 10.1016/j.chempr.2018.04.008
G.T. Venkanna, S. Tammineni, H.D. Arman, et al., Organometallics 32 (2013) 4656–4663. doi: 10.1021/om400630q
K.J. Jonasson, O.F. Wendt, Chem. Eur. J. 20 (2014) 11894–11902. doi: 10.1002/chem.201403246
C.L. Rock, R.J. Trovitch, Dalton Trans. 48 (2019) 461–467. doi: 10.1039/c8dt04608e
S. Chakraborty, J. Zhang, J.A. Krause, et al., J. Am. Chem. Soc. 132 (2010) 8872–8873. doi: 10.1021/ja103982t
J.Y. Yang, R.M. Bullock, W.J. Shaw, et al., J. Am. Chem. Soc. 131 (2009) 5935–5945. doi: 10.1021/ja900483x
M.L. Helm, M.P. Stewart, R.M. Bullock, et al., Science 333 (2011) 863–866. doi: 10.1126/science.1205864
U.J. Kilgore, J.A.S. Roberts, D.H. Pool, et al., J. Am. Chem. Soc. 133 (2011) 5861–5872. doi: 10.1021/ja109755f
J.Y. Yang, S.E. Smith, T. Liu, et al., J. Am. Chem. Soc. 135 (2013) 9700–9712. doi: 10.1021/ja400705a
Y. Li, J.-Y. Chen, Q. Miao, et al., J. Am. Chem. Soc. 144 (2022) 4365–4375. doi: 10.1021/jacs.1c08609
X. Zhai, M. Pang, L. Feng, et al., Chem. Sci. 12 (2021) 2885–2889. doi: 10.1039/d0sc06787c
M. Pang, C. Wu, X. Zhuang, et al., Organometallics 37 (2018) 1462–1467. doi: 10.1021/acs.organomet.8b00114
M. Pang, J.-Y. Chen, S. Zhang, et al., Nat. Commun. 11 (2020) 1249. doi: 10.1038/s41467-020-15118-x
H. Song, K. Ye, P. Geng, et al., ACS Catal. 7 (2017) 7709–7717. doi: 10.1021/acscatal.7b02527
X. Zhuang, J.-Y. Chen, Z. Yang, et al., Organometallics 38 (2019) 3752–3759. doi: 10.1021/acs.organomet.9b00486
J. Liu, H. Song, T. Wang, et al., J. Am. Chem. Soc. 143 (2021) 409–419. doi: 10.1021/jacs.0c11448
J. Liu, J.-Y. Chen, M. Jia, et al., ACS Catal. 9 (2019) 3849–3857. doi: 10.1021/acscatal.8b05136
P.L. Holland, M.E. Smith, R.A. Andersen, J. Am. Chem. Soc. 119 (1997) 12815–12823. doi: 10.1021/ja971830o
S. Park, S. Chang, Angew. Chem. Int. Ed. 56 (2017) 7720–7738. doi: 10.1002/anie.201612140
W. Meng, X. Feng, H. Du, Acc. Chem. Res. 51 (2018) 191–201. doi: 10.1021/acs.accounts.7b00530
X. Fan, J. Zheng, Z.H. Li, et al., J. Am. Chem. Soc. 137 (2015) 4916–4919. doi: 10.1021/jacs.5b03147
D.V. Gutsulyak, A. vanderEst, G.I. Nikonov, Angew. Chem. Int. Ed. 50 (2011) 1384–1387. doi: 10.1002/anie.201006135
C.D.F. Königs, H.F.T. Klare, et al., Angew. Chem. Int. Ed. 52 (2013) 10076–10079. doi: 10.1002/anie.201305028
F. Zhang, H. Song, X. Zhuang, et al., J. Am. Chem. Soc. 139 (2017) 17775–17778. doi: 10.1021/jacs.7b11416
X. Fan, J. Zheng, Z.H. Li, et al. J. Am. Chem. Soc. 137 (2015) 4916–4919. doi: 10.1021/jacs.5b03147
K. Nakaya, S. Takahashi, A. Ishii, et al., Dalton Trans. 50 (2021) 14810–14819. doi: 10.1039/d1dt01856f
S.R. Tamang, M. Findlater, J. Org. Chem. 82 (2017) 12857–12862. doi: 10.1021/acs.joc.7b02020

Figure 1 Structures of 1 (left) and 2 (right) with 50% probability thermal ellipsoids. Selected bond distances (Å) and angles (°): For 1 Ni−P 2.1558(7), Ni−O 1.886(2), Ni−Cp*(centroid) 1.752, P−Ni−O 89.21(6); for 2, Ni−P 2.1382(6), Ni−N 1.8848(18), Ni−Cp*(centroid) 1.744, P−Ni−N 87.21(6).

Figure 2 (a) Cyclic voltammograms of 1 and 2, and (b) solid-state structure of [1]BF4. Conditions: 1 mmol/L sample in THF, 0.1 mol/L nBu4NPF6; scan rate, 100 mV/s; potentials vs Fc+/0. Selected bond distances (Å) and angles: Ni−P 2.2307(6), Ni−O 1.8229(15), Ni−Cp*(centroid) 1.729, O−Ni−P 88.83(5)⁰.

Figure 3 Solid-state structure of [H1(Bpin)]. Selected bond distances (Å) and angle (°): Ni−H 1.42(2), Ni−P 2.0991(5), B−O1, 1.365(2), B−O2 1.367(2), B−O3 1.354(2); P−Ni−H 75.7(8).

Scheme 4 Ni(Ⅱ)-catalyzed hydroboration of ketones and imines. Reaction conditions: substrates (0.3 mmol), HBpin (0.45 mmol), catalyst 1 or 2 (6 µmol, 2 mol%) in C6D6 (0.5 mL). Yields determined by 1H NMR spectroscopy are based on tetraethylsilane (0.03 mmol) as an internal standard.

Scheme 5 Ni(Ⅱ)-catalyzed hydroboration of N-heteroarenes. Reaction conditions: substrate (0.3 mmol), 1 (6 µmol, 2 mol%), HBpin (0.45 mmol) in 0.5 mL C6D6. Yields were determined by 1H NMR spectroscopy using tetraethylsilane (0.03 mmol) as an internal standard. a No reaction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



 下载:
下载: